#body dystrophin
Text
Welcome to the latest episode of ‘Did I lose weight? Or did my clothes stretch out?’
0 notes
Text
There are multiple risk factors for the development of osteoporosis. These include but are not limited to advanced age, cigarette smoking, chronic glucocorticoid therapy, low body weight, previous fractures, history of rheumatoid arthritis, and excessive alcohol intake.
Antiepileptic drugs (AEDs) are associated with adverse effects on bone health. The AEDs most commonly associated with altered bone metabolism are those that induce the cytochrome P450 enzyme system. Specifically, the AED phenytoin has a direct inhibitory effect on intestinal calcium absorption and can stimulate osteoclastic bone resorption.
A score of less than -2.5 on DEXA scanning would indicate osteoporosis, a score of -1.0 to -2.5 would indicate osteopenia, and a score greater than -1.0 indicates normal bone density. Additionally, a fragility fracture (fracture from minor trauma such as a fall from a standing height or less) is diagnostic of osteoporosis without any further workup.
Bottom Line: The risk factors for osteoporosis include advanced age, cigarette smoking, chronic glucocorticoid therapy, physical inactivity, poor intake of calcium and vitamin D, body weight less than 127 lb, previous fractures, anticonvulsant use, hyperthyroidism, celiac disease, rheumatoid arthritis, and excessive alcohol intake.
The characteristic need for the child to use his hands to push himself to an upright position when arising from the floor is the Gower sign. It results from weakness in the proximal lower extremity muscles. Physical examination reveals pseudohypertrophy of the calf, lumbar lordosis, a waddling gait, shortening of the Achilles tendons, hypotonia, and hyporeflexia or areflexia. Patients usually use wheelchairs by age 12 or 13. Additional complications include delayed growth, dilated cardiomyopathy, increased fractures, progressive scoliosis with impaired pulmonary function, cognitive impairment, and eventual respiratory insufficiency.
Bottom Line: Elevated laboratory markers in the setting of Duchenne muscular dystrophy include serum creatine kinase, aspartate aminotransferase, and alanine transaminase.
COMBANK Insight: DMD is a progressive, myopathic disorder inherited in an X-linked, recessive fashion and caused by a defective gene on the X chromosome responsible for dystrophin production. Dystrophin normally serves to stabilize and prevent the breakdown of muscle fibers. Loss of dystrophin thus leads to muscle fiber degeneration, resulting in muscle weakness.
Wilson Disease, also known as hepatolenticular degeneration, is an autosomal recessive defect involving an ATPase, Cu2+ -transporting, β-polypeptide. In this disease state, copper is deposited in tissues of the brain, liver, kidneys, and Descemet membrane of the cornea (known as Kayser-Fleischer rings, which are seen in the exhibit). Parkinsonian-like tremor and dementia may be evident when the nervous system is affected. Renal tubular damage occurs in the kidneys and cirrhosis can develop in the liver. Diagnosis can be made with the observation of decreased serum ceruloplasmin. Treatment includes penicillamine, which creates a soluble complex with the metal that can be excreted through the urine.
Bottom Line: A Kayser-Fleischer ring is a golden brown ring observed in the Descemet layer of the cornea as a result of copper deposition and is seen in Wilson disease. Labs reveal low serum ceruloplasmin and treatment with penicillamine can stop the progression of this degenerative disease state.
The types of cancers that metastasize to the bone most commonly are lung, breast, thyroid, renal, and prostate. Multiple myeloma and lymphoma can both form lytic lesions from their origins within the marrow.
A SPEP and UPEP are sensitive for multiple myeloma and should be obtained along with a measurement of the patient's total protein and globulin levels.
Bottom Line: The initial workup of a new lytic lesion generally involves a CT chest/abdomen/pelvis, SPEP/UPEP, bone scan, basic labs, and mammogram or PSA
#osteoporosis#Duchenne muscular dystrophy#muscular dystrophy#Wilsons disease#multiple myeloma#cancer
3 notes
·
View notes
Text
DYNE-251 for the treatment of Duchenne Muscular Dystrophy Received FDA Fast Track Designation
HK-Magicure -- On October 31, Dyne Therapeutics announced that the U.S. FDA has granted Fast Track designation for DYNE-251 for the treatment of Duchenne muscular dystrophy (DMD) mutations amenable to exon 51 skipping. DYNE-251 is being evaluated in the Phase 1/2 DELIVER global clinical trial.

DYNE-251 is Dyne’s product candidate being developed for people living with Duchenne muscular dystrophy (DMD) who are amenable to exon 51 skipping. DYNE-251 consists of a phosphorodiamidate morpholino oligomer (PMO) conjugated to a fragment antibody (Fab) that binds to the transferrin receptor 1 (TfR1) which is highly expressed on muscle. It is designed to enable targeted muscle tissue delivery and promote exon skipping in the nucleus, allowing muscle cells to create a truncated, functional dystrophin protein, with the goal of stopping or reversing disease progression.
In preclinical studies, robust and durable exon skipping and dystrophin expression were observed in the mdx mouse model in skeletal and cardiac muscle as well as reduced muscle damage and increased muscle function. In non-human primates, DYNE-251 demonstrated a favorable safety profile.
DELIVER is a Phase 1/2 global clinical trial evaluating DYNE-251, consisting of a 24-week multiple ascending dose (MAD) randomized placebo-controlled period, a 24-week open-label extension and a 96-week long-term extension. The trial, which is designed to be registrational, is expected to enroll approximately 46 ambulant and non-ambulant males with DMD who are ages 4 to 16 and have mutations amenable to exon 51 skipping therapy. The primary endpoints are safety, tolerability and change from baseline in dystrophin levels as measured by Western blot. Secondary endpoints include measures of muscle function, exon skipping and pharmacokinetics. Dyne anticipates reporting data from the MAD placebo-controlled portion of the DELIVER trial on safety, tolerability and dystrophin in the second half of 2023.

About Duchenne Muscular Dystrophy (DMD)
Duchenne Muscular Dystrophy (DMD) is a rare disease caused by mutations in the gene that encodes for dystrophin, a protein critical for the normal function of muscle cells. These mutations, the majority of which are deletions, result in the lack of dystrophin protein and progressive loss of muscle function.
DMD occurs primarily in males and affects an estimated 12,000 to 15,000 individuals in the U.S. and 25,000 in Europe. Loss of strength and function typically first appears in pre-school age boys and worsens as they age. As the disease progresses, the severity of damage to skeletal and cardiac muscle often results in patients experiencing total loss of ambulation by their early teenage years and includes worsening cardiac and respiratory symptoms and loss of upper body function by the later teens. There is no cure for DMD and currently approved therapies provide limited benefit.
For more articles on medicines, click here: hkmagicure
Hong Kong Magicure Medical Center has long been focusing on the import and export of new drugs, special drugs and rare disease drugs in the field of oncology. Welcome to inquiry: [email protected].
3 notes
·
View notes
Text
Ugh, hate that like. Nothing makes sense.
Because. "Gangliated" means to have ganglia nerve cells. "Utrophin" is a strange choice, its big brother "Dystrophin" is a structural protein that muscle cells need to survive - utrophin occurs in usually only in human fetuses and is replaced, except in the cases of muscular dystrophy. "Immuno-," just means it relates to the immune system, "Latency" refers to a dormant period inside the host. Together it means that the immune system creates it, which is does, we'll see more of later. Finally, "Toxin" means it is produced by a living organism such as a bacteria or a virus (though viruses are considered nonliving, the one exception). Compare to a "Toxicant," which is something man-made. There is a difference.
In short, GUILT is a toxin that, when injected into a host, tricks the body into synthesizing its own personalized killing machines.
But. That's a virus. That's virus behavior. So it's just. What the hell? My best guess is that it could just be a toxic solution carrying viral DNA. basically the exact opposite of a vaccine. Which means.. The first instances of GUILT could be transferred through fake vaccines. Eidoth Pharmaceuticals is a medical company, they could probably get away with making a shot that gives you GUILT. Hooray.
THAT, and imagine all the anti-vaxxers up in arms because "Oh, this vaccine gave me a deadly virus!" It'd be a fucking nightmare.
4 notes
·
View notes
Text
The Work of CureDuchenne

CureDuchenne is a nonprofit dedicated to saving the lives of boys with Duchenne muscular dystrophy (DMD). DMD belongs to a category of medical conditions called muscular dystrophies, a group of genetic disorders where muscles experience weakness. DMD complicates even the most basic activities. In addition, as the condition progresses, the muscles responsible for heart and breathing functions are also affected, further weakening the body.
This medical condition mostly affects boys. By age 12, most of them start to experience social isolation. Studies report that most boys with DMD do not make it to their late twenties, and those who do are usually confined to a wheelchair. There are about 20,000 boys living with DMD in the US, and more than 300,000 worldwide.
CureDuchenne is a renowned global innovation, research, and patient care organization committed to developing medical solutions that can improve and extend the lives of boys living with DMD. Together with its board of directors and scientific advisory board, the organization’s activities include extending patient ambulation in partnership with healthcare and medical professionals and pharmaceutical companies. Ambulation refers to the ability to walk without needing any assistance.
CureDuchenne’s diverse efforts include focusing on DMD treatments, investing in companies pursuing dystrophin-restoring and anti-inflammatory approaches. Dystrophin is the protein that stabilizes muscles.
The organization also seeks innovative technologies to overcome current therapy limitations. CureDuchenne Ventures’ investments have attracted over $3 billion in financing, leading to successful exits with venture capital, biotech, and pharmaceutical partners.
0 notes
Text
Advancements in Duchenne Muscular Dystrophy Treatment Market: A Year in Review
Introduction:
As we celebrate the first year since your inception, it's crucial to reflect on the progress made in the medical field, particularly in the realm of Duchenne Muscular Dystrophy (DMD) treatment. Duchenne Muscular Dystrophy is a rare and devastating genetic disorder that primarily affects young boys, progressively leading to muscle degeneration and loss of motor function. Over the past year, the Duchenne Muscular Dystrophy Treatment Market has witnessed significant advancements, offering hope to patients and their families.
Current Landscape:
DMD is caused by a mutation in the dystrophin gene, leading to the absence or deficiency of the dystrophin protein, vital for muscle integrity. Historically, DMD lacked effective treatment options, primarily focusing on symptom management and supportive care. However, the landscape is rapidly evolving, with innovative therapies emerging to address the root cause of the disease.
Gene Therapies:
One of the groundbreaking developments in the past year involves gene therapies targeting the dystrophin gene. These therapies aim to either replace or repair the faulty gene, offering a potential cure for DMD. Several clinical trials have shown promising results, with some gene therapies demonstrating significant improvements in muscle function and mobility.
Exon Skipping Therapies:
Exon skipping therapies, another area of active research, focus on modifying the RNA produced by the dystrophin gene. By skipping specific exons, these therapies aim to restore the reading frame, allowing the production of a truncated but functional dystrophin protein. Recent trials have reported encouraging outcomes, showcasing the potential of exon skipping as a viable treatment strategy.
Emerging Pharmacological Approaches:
Pharmacological interventions continue to play a crucial role in DMD treatment. Novel drugs targeting inflammation, fibrosis, and muscle regeneration are undergoing clinical trials. These therapies aim to alleviate symptoms, slow disease progression, and enhance patients' overall quality of life. The diverse range of pharmacological approaches reflects a comprehensive strategy to tackle the complex nature of DMD.
Collaboration and Regulatory Support:
The past year has witnessed increased collaboration among pharmaceutical companies, research institutions, and regulatory bodies. This collaborative effort has accelerated the development and approval processes for DMD therapies. Regulatory agencies have recognized the urgent need for effective treatments, leading to expedited review processes and approvals for promising therapies.
Challenges and Future Outlook:
While the advancements in the Duchenne Muscular Dystrophy Treatment Market are undoubtedly promising, challenges persist. Accessibility and affordability of these cutting-edge therapies remain critical concerns. Additionally, long-term safety and efficacy data are essential to ensure the sustained benefits of these treatments.
Looking ahead, ongoing research and clinical trials will likely bring forth further innovations in DMD treatment. The integration of personalized medicine and a deeper understanding of the genetic variations contributing to DMD will pave the way for more targeted and effective therapies.
Conclusion:
As we mark the first year in the journey toward conquering Duchenne Muscular Dystrophy, the progress made in the treatment landscape is both inspiring and hopeful. The collaboration between researchers, pharmaceutical companies, and regulatory bodies reflects a shared commitment to transforming the lives of those affected by this devastating disorder. The year ahead holds the promise of continued advancements, bringing us closer to a future where Duchenne Muscular Dystrophy is not just manageable but ultimately curable.
0 notes
Text
Muscular Dystrophy: What You Need To Know?
If your or your child’s muscles are becoming weak and less flexible over time, you better get a check-up done by a doctor for muscular dystrophy.
A muscular dystrophy is a group of diseases that affects the muscles, and only if you lack this problem in your genes will your muscles continue to be strong. The condition may show symptoms early in childhood for some people, while others do not have symptoms until they reach their teenage years or their middle age.
There are various types of muscular dystrophy, and each is distinct according to the following:
The genes that create it,
The affected muscles,
The person’s age when the first symptoms appeared,
How quick is the progression of the disease?
Here are 9 of the major forms of this disease:
* Duchenne muscular dystrophy is the most common type and mostly affects boys. The symptoms start between ages 3 and 5.
* Becker muscular dystrophy is the milder form of Duchenne, with symptoms starting between ages 11 and 25.
* Myotonic is the most common form in adults.
* Congenital muscular dystrophy occurs at birth or early infancy.
* Limb-girdle muscular dystrophy causes weakness and wasting of the muscles in the arms and legs.
* Facioscapulohumeral muscular dystrophy affects the muscles of the face, shoulders, upper arms and lower legs.
* Distal muscular dystrophy is characterised by wasting and weakness of voluntary distal muscles, which are farther from the centre of the body, such as lower arms, hands, legs and feet.
* Emery-Dreifuss muscular dystrophy affects the skeletal muscles and the heart.
Muscular dystrophy symptoms usually appear either during childhood or in the teen years. Generally, a child with this disease:
- frequently loses balance and collapse
- have weak muscles
- experience muscle cramps
- have difficulty rising from the bed, climbing stairs, jumping or running
- walk with short steps and in clumsy, swaying motion; walk on their toes
- have curved spine (scoliosis)
- have eyelids hanging down limply
- have heart problems
- experience difficulty in breathing or swallowing
- have vision problems
- feel weakness in the muscles of the face
Currently, no clear muscular dystrophy treatment is yet to cure the disease. However, many treatment plans and interventions could help the patient improve their symptoms, thus making life easier for the child and the family.
Here are some doctor-recommended treatments, the application of which should be based on what type of muscular dystrophy a person is suffering from.
1. Physiotherapy (physical therapy) uses different exercises, including stretches, to keep the muscles strong and flexible.
2. Occupational therapy teaches the patient to make the most use of what their muscles can do. Also, they will learn to use wheelchairs, braces and other devices to aid them in their daily functions.
3. Speech therapy trains the person with speech and language problems due to a weak throat and facial muscles to speak more clearly.
4. Respiratory therapy helps restore or improve lung functions; helps with difficulty breathing.
5. Medications to help ease symptoms, such as:
* injection medication to help increase dystrophin production
* anti-seizure drugs to help lessen muscle spasms
* medications for blood pressure to help with heart problems
* immunosuppressant drugs to slow down the damage to muscle cells
* steroids to slow down damage to muscle cells; also help the patient to breathe easily
6. Surgery can help with various muscular dystrophy complications, like trouble swallowing or heart problems.
Although MDs of any type cannot be fully cured, any of the above lists can be successful treatments of muscular dystrophy (therapies, injections, medications, surgery). They can help lengthen the time a child or adult with the disease can remain mobile. They also help strengthen the patient’s heart and lung muscles.
It is quite difficult for parents to see their child lose their muscle strength and be unable to do things other children can. Managing muscular dystrophy is challenging, but don’t let it stop your child from enjoying life.
If you have questions or need more information about muscular dystrophy, please contact Mission Walk today. You need to fill out our online contact form and send it, and one of our team members will be happy to get back to you.
#Muscular dystrophy symptoms#Muscular dystrophy treatment#Successful treatment of muscular dystrophy#muscular dystrophy
1 note
·
View note
Video
cas: 1621169-52-5 ACE-031 Myostatin inhibitory peptide 7
ACE-031 is a soluble form of the type IIB activin receptor (ACVR2B), which is a potent regulator of muscle growth in vivo. Myostatin, a negative regulator of muscle growth, is bound by ACVR2B and subsequently inactivated. Research in mouse models shows that knocking out the activin receptor boosts mass in all muscle cells. As a result of this connection, ACE-031 has been investigated in several clinical trials as a potential treatment for muscle-wasting diseases like Duchenne muscular dystrophy (DMD). Activin receptors also play important roles in the development of gametocytes, particularly sperm. The receptor is also often found to be inactivated in prostate cancer and is damaged or dysfunctional in several different types of colorectal cancer.
What Is ACE-031?
ACE-031 is a soluble form of the activin type IIB receptor and is known to bind to myostatin and neutralize its effects. Myostatin is a protein, produced by muscle cells, that inhibits both hypertrophy and hyperplasia of skeletal muscle. ACE-031 also likely has effects on bone metabolism, fat storage, and the health of sperm.
ACE-031 Research and Muscle Protection After Menopause
ACE-031 was tested in a small clinical trial to determine if it could help to maintain muscle mass in postmenopausal, healthy women. Maintaining muscle is critical to long-term bone health and helps to reduce the rate of joint injuries, falls, etc. in both men and women as they age. In a small placebo-controlled trial, ACE-031 produced significant increases in both lean body mass and thigh muscle volume after just a single dose. Results were observed 29 days after the injection[1]. The study was particularly interesting because a secondary, unexpected outcome was observed. Trials participants receiving ACE-031 showed improvement in serum biomarkers of both bone and fat metabolism. This suggests that ACE-031, while primarily associated with muscle growth, likely inhibits fat storage and boosts bone production.
ACE-031 May Be Necessary for Maximum Muscle Cell Growth
Research in mice indicates that maximal skeletal muscle growth can only be attained with a myostatin inhibitor like ACE-031. Furthermore, it appears that blockade of myostatin by multiple avenues is most beneficial[2]. While these findings are preliminary, they suggest that maximum muscle protection in muscle-wasting conditions might require a multifaceted approach utilizing therapies that both boost growth (growth hormone, IGF-1) and reduce muscle wasting (ACE-031).
ACE-031 Research and Strength
There appears to be more to the ability of ACE-031 to improve muscle function than inhibition of myostatin. Research in mice indicates that ACE-031 can improve force-generating capacity in muscle tissue, in part by preserving energy supply and shifting muscle thermodynamics toward oxidative respiration. In the mice, administration of ACE-031 improved maximum and total contractile force by 40% and 25% respectively. There was no overall change in fatigue of muscle though, indicating that ACE-031 improves muscle strength without affecting energy dynamics. The research revealed no changes in ATP homeostasis or contractile efficiency[4].
ACE-031 Tested in Clinical Trials for Muscle Repair
Duchenne muscular dystrophy (DMD) is an x-linked recessive condition characterized by severe muscle loss. It generally leads to an inability to walk by age 12 with muscles that are lacking in protein content but that are exceptionally high in fat content. Due to a dysfunctional dystrophin protein, muscle cells in DMD are weak and prone to damage. Though this is the primary cause of the disease, a secondary effect occurs when myostatin leaks from damaged muscle cells and slows or inhibits growth in other cells. Gene therapy to address the dystrophin dysfunction has so far proved untenable, but there is currently hope that ACE-031 can slow the muscle damage by reducing the secondary impact caused by myostatin.
In a recent clinical trial, subcutaneous injection of ACE-031 every 2-4 weeks
0 notes
Text
When you have probably the worst body compared to all her exes but your love for her is unmatchable so whenever you think of it you’re like “Is she really attracted to me physically or does she only pretend she is and actually hate our intimate moments?”
Ehe
#cries in ugly#glob bod gang#squishier than a pillow#overthinking#smolbean’s vent#why does she even love me#unattractive#somebody switch your body with mine#too depressed to workout#never satisfied with my body#cries in body dystrophin#wlw#wlw ldr#wlw blog#wlw love#lesbian poetry#sapphic poetry#wlw aesthetic#wlw yearning#girls who like girls#sappho
41 notes
·
View notes
Text
TAFAKKUR: Part 317
FROM CELL TO BODY
Living creatures, which are arranged in an organization by a hierarchic system of non-living matter, are combinations of syst eachms within other. Each level contains unique systemic arrangements. However, in different levels, each organization works in a wonderful and perfect harmony with other levels so that the living creature survive. As a result, the responsibilities entrusted its structure and function(s) are not interrupted.
WHO CREATED SUCH MIRACULOUS ENTITIES?
At present, many highly educated people and scientists maintain there should be a creator who knows and has the power to design of all these perfectly working organizations. Starting from an atom's particles, this systemic structure consisting of atoms, molecules, macromolecules, cell organelles, cells, tissues, organs, systems, and finally a living creature exists in all ecological balances on Earth. Without seeing the impact of such a wide and large power, it is not logical to expect that all that we see in creation is the result of unconscious nature and thus conclude that everything occurred by blind happenstance.
We remain alive because millions of cells work together. Each maintains its life by being perfectly divided into units and having complex mechanisms within its structure. Each unit has unique features. When these units come together, large and unpredictable new features might occur. For example, hydrogen is flammable and oxygen is burning, but when they join together to form water, there is no fire.
A cell, which consists of electrons, protons, and neutrons, might function differently according to its location in the body. When cells are combined with each other, they form cell groups that acquire such characteristics as flexibility, movement, reproduction, and nerve transmission. We know that all such body units as cells, tissues, and organs have a perfect integrated system. And yet it is still unclear how they combine and carry out the functions necessary to make eyes, the brain, bones, muscles, and so on, all of which are composed mainly of carbon, hydrogen, oxygen, nitrogen, and phosphate.
In short, many scientists are very curious about how brain tissues can perform hundreds of different functions (e.g., hearing, thinking, and feeling) even though they are composed of atoms.
RECENT DISCOVERIES
For the last two decades, scientists have been working on small molecules, DNA, RNA, and proteins, in order to shed some light to the unknown aspects of life. The Human Genome Project (HUGO) is one of the largest projects and is scheduled to be finished by 2005. Its goal is to sequence the cell's genetic material, which presumably numbers some 100,000 genes, and explain each one's function(s). However, so far only 10,000 to 20,000 are known. And this despite the fact that scientists have spent centuries learning how molecules are formed and what kind of force drives the atoms to form molecules!
Several famous foundations, among them Yale University, New York's Cold Harbor Laboratory, and Germany's Max Planc Institute, have been researching questions related to the body's structure. For example, how did it occur in such a small living cell, even though its building mechanism and systems are far more complicated than the system, which is all around us? How are dead cells expelled and replaced by new ones without disrupting any system's continued functioning.
In short, living creatures still hold many secret--so many, in fact, that despite recent technological advancements many unknown subjects remain. What we know may be compared to a drop of water and the ocean.
DNA AND GENES
Excluding blood cells, one body contains millions of cells. For instance, there are approximately 5 million white blood cells within 1 mm3 of blood. Each cell has a nucleus (its brain), and each nucleus contains genetic codes (DNA). Such genetic information as hair and eye color, bodily height, and blood group is hidden within the folded DNA and packed onto 46 human chromosomes distributed. If we could unfold the DNA package within each cell, we would get a 2-meter long DNA strain.
In addition, each chromosome contains small DNA units (genes). Scientists estimate that there are about 100,000 genes, each of which has a unique function, in a human genome. For instance, there are globin (the major component of hemoglobin) genes on chromosomes 11 and 16, and individual genes on the X chromosome that enable us to see colors. Another gene on the X chromosome synthesizes a dystrophine protein within our muscles, other genes on chromosome 6 control ferine metabolism, another gene on chromosome 7 controls the ion traffic, and so on.
When we look at a gene's structure, we realize that it they only are encoded by four letters (nucleotides): adenine (A), guanine (G), cytosine (C), and timin (T). The different orders of these four nucleic acids enable the gene to encode billions of protein amino acid combinations. The smallest gene, globin, is composed of 60,000 letters; the longest gene, distrophine, is composed of 2.4 million letters. If any letter is missing or is in the wrong place within the globin gene (e.g., AATG_ letter missing or AATTC letter change instead of AATGC), the ensuing point mutation, deletion, or insertion within the original order causes serious and incurable genetic diseases.
Many mutations occur within the functional parts of the 100,000 genes; however, they are instantly (10-10 second) repaired by the DNA 's repair mechanism. If not, serious diseases might occur. Interestingly, not all mutations cause genetic diseases, for while one mutation could be common within one human population, it might not occur so frequently within another one. Amazingly, our genes, composed of billions of letters (but only from A, T, C, and G) and always work in a harmony and collaboration so that our body can survive in a perfect manner. Therefore, spontaneous mutations or breakdowns are instantly repaired and old cells are replaced by new ones so quickly that we are not even aware of it.
CONCLUSION
In sum, the Creator has given each living entity a perfect and aesthetic body that functions in a most amazing manner. The only thing we have to do is thank the Creator for His generosity. That is perhaps the easiest task for us to do.
#allah#god#prophet#Muhammad#quran#ayah#sunnah#hadith#islam#muslim#muslimah#hijab#help#revert#convert#religion#reminder#dua#salah#pray#prayer#welcome to islam#how to convert to islam#new convert#new revert#new muslim#revert help#convert help#islam help#muslim help
2 notes
·
View notes
Text
Pidge is actually trying to take this a tiny bit seriously. Last night, while she was working on moving her work station into the makeshift pharmaceutical laboratory she’s set up for herself over the last few months, she was putting together a presentation, like it’s a business pitch or a grant funding exercise. Still, it’s easier to illustrate her point when she has diagrams to go off of. The fancy little holograms from her PADD can even be manipulated in real-time in three-dimensional space, for added cool factor.
It also means she can keep her thoughts together as she goes through the theoretical aspects of this with @swordsedge Ulaz. Before she begins, she takes a shot glass, fills it with the Olkari root extract she’s come to love so much, and knocks it back like it’s so much liquor. That should keep her going for the next eight or so hours and stave off the fluorescent-inspired headache she’s guaranteed to get if she works down here too long. She offers some to the Galra in front of her, but he declines. Reasonable. He doesn’t know what it is, and she could have tampered with it, so she’s not offended.
They’d had a brief conversation last night, as well, about how to structure this upcoming week. Pidge had asked Ulaz what the Galra Empire would do for someone who had a genetic degenerative disease. The answer, unsurprisingly, was a mercy cull. For an empire driven by expansion at all costs, a disabled life is not one that can be afforded. Ulaz did show the correct amount of disgust as he explained, at least, which reassured Pidge that he was here for the right reasons, to do the right thing. What wasn’t so reassuring was that he hadn’t actually encountered this specific problem before, as a medical officer.
Tilting her PADD against her empty glass so the holograms can project onto the table, Pidge launches into her explanation. “so, you understand what we need to do here,” Pidge reminds Ulaz. “this is different from just keeping shiro in stasis and keeping disease from progressing. this is total genomic overhaul.” She flicks the first diagram out from her screen to the table, starts spinning it--a puffy little X shape made of squiggles. “what we’re working with is the x chromosome, a location on the short arm called p21.2-p21.1.” When she zooms in with her fingers, there's a noticeable length difference between the two top arms of the chromosome. “there’s a deletion here--not one of the worst, but not in a good place, either. this codes for dystrophin: the protein that builds human muscle. without it, the muscles we’re born with can’t be effectively re-built when they’re damaged. usually, you’d have a backup on the other matching pair in your chromosome set, so your body could just use the one that works and ignore the one that doesn’t, but shiro can’t do that, because he doesn’t have a second x chromosome, he has a y chromosome. which, don’t tell it i said this, but it’s pretty useless, aside from sry. poor little thing. smallest in the human genome.”
This is probably stuff Ulaz already knows. Based on what Pidge surmises about Galra, just from pure conjecture surrounding the fact of Keith’s existence, they also must have a similarly-based biology, with double-helix DNA, ACGT pairs, X/Y sex chromosomes, even the same number and arrangement of chromosomes. Otherwise, Galra wouldn’t be able to reproduce with humans, or proliferate so far with so many other alien races. Still, it helps to start from the common denominator and build up to more complex premises.
Pidge pinches her fingers together, then spreads them to zoom in on her DNA diagram--to the portion that’s missing. “there’s maintaining the dystrophin shiro still has, and there’s teaching his body how to make it for himself. two different things. he already had weakness in his legs, to be expected, but now you’re telling me he’s having trouble breathing. that means his diaphragm can’t repair itself. he’s too weak to work his own lungs. that’s... that’s advanced. the only way it could be worse is if it was in his heart, and we don't know that it's not. so, we can’t just plug this with pharmaceutical intervention. giving him the actual dystrophin protein isn’t, by itself, going to get him where he needs to go. he needs to do it for himself, and he needs to be able to rebuild what’s been lost on top of it. that means...”
Another diagram flicks next to the first. This one's the clip of what's missing. “i have to get this, here, but... everywhere. as far as you're telling me, this is something the galra weren’t even interested in devoting resources to. it’s something humans haven’t quite been able to achieve, even with crispr, our most advanced gene splicing engineering technology. altean alchemy isn’t suited to this, and i can’t see that they've ever attempted a genetic cure, just an amino acid replacement. the olkari seem to find it anathema to attempt it, even with their advanced biohacking abilities. but i’m--we’re not dealing with just one set of medicine. we’re not limited here. i can use all of this accumulated knowledge and make something bigger than the sum of its parts. i just need to run this by you, theoretically speaking, to see if it’s even possible in practice.”
Dismissing the first diagram to focus on the second, she twists her two hands, pulls them apart, and it zooms in on the individual molecules making up the DNA helix: red adenine paired with green thymine, yellow cytosine paired with blue guanine, clumped in threes (that’s a slight liberty with the illustration, but it works for these purposes). “coran’s taught me how to use this lab to make pharmaceutical compounds i thought would be impossible with the materials we have. apparently all you have to do is ask these atoms and molecules nicely to create their bonds. so far i’ve been... moderately successful in using it.” That’s false modesty. Pidge has been able to synthesize a full medication line for Shiro by now, from advanced corticosteroids to muscle relaxers, from gene-targeted therapies to painkillers. “but, i mean, dna is just a bunch of molecules, when you get down to it. huge, snarled-together molecules, but molecules all the same. the backbone of the helix is the same. the a, c, g, t are the same. if i can teach the lab to make the individual components, it’s just an issue of putting the building blocks in the right order and making them stick together. that part, actually making the gene i need, that’s the part i have the most confidence in. i know i can do it. what i don’t know is how much time it’s going to take, or if i can accelerate it by redirecting non-essential ship power to this one resource. and i won’t know for sure until i get started on it. but, the good news is, i know what i need to make and how i need to make it. easy.” Relatively speaking, of course.
The next image Pidge pulls up is entirely new. “this--this part’s more complicated. this little device is crispr. technically it’s a repeating genome sequence that humans synthesized from a bacteria, but you can use it for genome modification. depending on what kind of rna you attach to it, you can use it to snip out genes entirely, or cut and paste from one mis-transposed location to another. notice i didn’t say insert. it needs to get the material from somewhere to insert it in the first place, and creating the right sequence out of nothing was always a little too difficult to stabilize in human trials. plus, there were ethical concerns with using it on stem cell lines. no such worries here. if i use altean alchemy to create the missing piece, and if i use the right rna to point it at xp21.2 through .1, it should plunk it right into place. and there’s no medico-ethical dilemma present for doing this with a full-grown person, like there would be if we were trying to fix it in a zygote. it doesn’t even generate the should-we argument. now, getting the rna to target the right location, and getting the delivery mechanism to be stable, and getting it to lock into place, that’ll be a little more difficult.”
What flashes into the set of images Pidge is using, this time, is a series of ones and zeroes. “that's where the olkari technology comes in. their tiaras use human brainwaves, sent as binary code, to modify messenger rna, to redirect plants on what genes they should be expressing at any given time. it unlocks a gene’s potential. this should be the key to not only targeting the right location for the gene insertion, but also in making sure that it’s getting used correctly to code for dystrophin. the question you’re probably about to ask is, how does this work with dna when dna isn’t written in binary? but it’s not about reading it, it’s about finding it. rna will read it for itself, pull the correct amino acids, and make dystrophin. cells are pretty smart that way.”
Dismissing all those prior symbols, Pidge finally pulls up a diagram of the human body. “so, congratulations. using a series of increasingly unstable chemical reactions pulling from the most advanced medicine, science, engineering, and coding from three different starfaring species, we created, spliced in, and activated exactly one copy of the dystrophin-coding gene, into one cell.” The hologram zooms in to some generic muscle strand of the forearm. “that cell could die before undergoing mitosis. even if it survives, that’s no guarantee that the new, fixed genome will propagate very far, even within the same physical location of the body.” A red flash, indicating failure.
“but, if i’m understanding your research correctly, there’s something you can do with filtered quintessence to not just make it stick around, but to get it to actually change the whole body genome. this is the part that i’m the most skeptical about,” just in case Ulaz couldn't tell from her tone. “i don’t know how quintessence works at the best of times. as far as i care, though, if it does what you say it will, then it can be literal space magic--as long as it works by a set of fixed principles. if you’re saying we can wash out the old genome and, i guess, dye the new one into place by steeping shiro in enough quintessence, it’s worth a try.”
Presentation over. Pidge collapses her diagrams, puts her PADD face-down on the table. When she catches Ulaz’s face, his expression is unreadable. Just like always, really. “so, after all that, i have two questions for you. one, does that sound like something we can, theoretically, even do? i don’t want to waste time or energy on research if it’s not going to pan out in real life. and even if it does, question number two, how much quintessence would it actually take to do something like that? are we talking on the level of a d-cell battery, car battery, aircraft engine, starship-class balmera crystal, the type of energy it would take to hold strand in stasis for eons--what do we need, and can we actually get it?”
7 notes
·
View notes
Text
little dmd issues shiro's had all his life that vary from childhood to teenagerhood to adulthood, progressively:
not being able to move/run/etc. quite as fast as other people or as well as he should without getting sore and/or winded
having to sometimes walk his hands up his legs to stand
having to use shock braces (not relevant anymore in middle adulthood) and two corticosteroids
having to take a beta blocker
having to use a bipap on and off (recently) because of a weakening diaphragm muscle
having a hard time eating enough, which is why he prefers hearty, calorically dense meals
stairs and putting on socks sucks, as does slippery feet/tripping when standing up and all the expected clumsiness therein
self-taught physical therapy is a thing (whereas now he has medical officers to assist with it if necessary)
exhaustion, physically mentally and socially; physical exhaustion takes it out of him
the fact that he's lived past 25 is unusual
sometimes when he takes a fall he can't get up right away
before the galra experimented on him he was very closely approaching quick deterioration. though he was in peak condition, he still needed to use braces to help relax his arm muscles. had this progressed it would have continued to deteriorate his other muscles and he would have had to chair himself
what quintessence binded surgery is going to do for him is inject quintessence + handmade serum into his body to, essentially, handicap dmd from killing him. when his muscles begin deteriorating, quintessence will regenerate them. however, this is give and take, and he will still have to live with all his symptoms, whether he likes it or not. it is not a cure, dmd is a genetic disease, and this is simply an aid that works much better than steroids do.
rebuilding/recooking replacement levels of dystrophin is going to be... much harder. pidge is injecting the gene she made into 12 of shiro’s diaphragm cells. the quintessence washout they’re going to do is going to wash out the impure quintessence from his arm and body and spread that genomic change so they don’t have to edit one cell at a time.
shiro still has to make baseline levels plus extra levels of dystrophin himself (his current handicap due to said genetic disease, dmd) and continue doing so with his new arm. quintessence requires one’s life force to use, so he’s going to have to have supplemental quintessence and serum injections for the next couple of days (we’re playing catchup here, get what i’m saying?), otherwise he’s going to be extremely ill.
and since pidge and ulaz don’t know how quintessence works entirely, this is a trial that has promising results, but is still dangerous.
shiro’s going to have to be watched for negative side effects. all of them.
5 notes
·
View notes
Text
Going through questions:
The genetic code is degenerate because more than one codon can code for 1 amino acid. Every tRNA goes with a specific amino acid; due to the degeneracy of the genetic code, the tRNA will respond to whichever codons go with its specific amino acid. The wobble hypothesis is that the third nucleotide doesn't have to do traditional base pairing.
Bloom syndrome is an autosomal recessive mutation in the BLM gene. It causes defect of helicase and presents with growth retardation, photosensitivity, immunodeficiency, and microcephaly.
Lyonization = X inactivation; in females, one of the X chromosomes is methylated and becomes a Barr body. This inactivated X chromosome becomes tightly would heterochromatin that isn't expressed. Heterochromatin = methylated DNA +deacetylated histones. I recall from listening to OnlineMedEd that acetylation makes DNA accessible. So deacetylation makes DNA less accessible. DNA is wrapped around histones. Methylation of DNA and deacetylation of histones makes the DNA less accessible for transcription. Euchromatin is not methylated and is easily accessible.
Precursor mRNA (pre-mRNA, aka heterogeneous nuclear RNA, hnRNA) is processed before leaving the nucleus--it gets the 5' cap and poly A tail added and introns are spliced out. Once all that happens, it's mature mRNA, which can leave the nucleus. There are also these things called P bodies that regulate mRNA in the cytoplasm. P bodies are involved in mRNA decay.
Uniparental disomy = the offspring receives 2 copies of a chromosome from 1 parent, and no copy from the other parent; leads to issues with imprinting. Uniparental disomy-> improper imprinting-> Prader-Willi and Angelman syndromes.
Down syndrome is often due to nondisjunction in meoisis, but can also be due to unbalanced Robertsonian translocations (you get too much of one copy of a gene and not enough of the other--this one I took detailed notes about from OnlineMedEd videos in my red notebook; if Robertsonian translocation causes Down syndrome, the pt will actually have a normal number of chromosomes [46], but the amount of genetic information from chromosome 21 will be more than it should be on the chromosomes the pt has, so it presents like there is an extra chromosome 21) or mosaicism (nondisjunction in mitosis causes some cells to have an extra chromosome 21, but not all cells).
Gowers’ sign is when pts with Duchenne muscular dystophy use their arms to get up; looks like they use their arms to “walk up” their own bodies. From Wikipedia:
Gowers' sign is a medical sign that indicates weakness of the proximal muscles, namely those of the lower limb. The sign describes a patient that has to use their hands and arms to "walk" up their own body from a squatting position due to lack of hip and thigh muscle strength.
Duchenne muscular dystrophy is an X-linked recessive mutation in the dystrophin gene. Frameshift and nonsense mutations cause shortened dystrophin gene. In unaffected people, dystrophin links with actin for support of glycoproteins in the plasma membrane of skeletal muscle cells. Defective dystrophin-> breakdown of sarcolemma, degeneration of muscle fibers, calf enlargement (it's really not the calf muscles that are enlarged--it's fat resulting from breakdown of the muscles), increased serum creatine.
Huntington disease is due to CAG trinucleotide repeats in the HTT gene. The more of these there are, the earlier the disease comes on and the more severe it is, which is called anticipation. Huntington's presents with chorea, depression/aggression/apathy, and dementia. Friedreich ataxia, fragile X syndrome, and myotonic dystrophy are also trinucleotide repeat diseases.
From Wikipedia:
Friedreich's ataxia (FRDA or FA) is an autosomal recessive genetic disease that causes difficulty walking, a loss of sensation in the arms and legs and impaired speech that worsens over time. Symptoms generally start between 5 and 20 years of age. Many develop hypertrophic cardiomyopathy and will require a mobility aid such as a cane, walker or wheelchair in their teens. As the disease progresses, people lose their sight and hearing. Other complications include scoliosis and diabetes mellitus.
FRDA is an autosomal recessive disorder that affects a gene (FXN) on chromosome 9 which produces an important protein called frataxin.[5]
In 96% of cases the mutant FXN gene has 90–1,300 GAA trinucleotide repeat expansions in intron 1 of both alleles.[6] This expansion causes epigenetic changes and formation of heterochromatin near the repeat.[5] The length of the shorter GAA repeat is correlated with the age of onset and disease severity.[7] The formation of heterochromatin results in reduced transcription of the gene and low levels of frataxin.[8] People with FDRA might have 5-35% of the frataxin protein compared to healthy individuals. Heterozygous carriers of the mutant FXN gene have 50% lower frataxin levels but this decrease is not enough to cause symptoms.
The condition is caused by mutations in the "FXN" gene on chromosome 9. The FXN gene makes a protein called frataxin. In FRDA, the patient produces less frataxin. Degeneration of nerve tissue in the spinal cord causes the ataxia; particularly affected are the sensory neurons essential for directing muscle movement of the arms and legs through connections with the cerebellum. The spinal cord becomes thinner, and nerve cells lose some myelin sheath.
No effective treatment exists, but there are several therapies in trials. FRDA shortens life expectancy due to heart disease, and some people can live into their sixties or older.
FRDA affects 1 in 50,000 people in the United States and is the most common inherited ataxia. Rates are highest in people of Western European descent. The condition is named after the German physician Nikolaus Friedreich, who first described it in the 1860s.
Homeobox (HOX) genes code for transcription regulators. A homeobox is highly conserved DNA of 180+ nucleotides. Mutations in homeobox genes lead to limbs in the wrong place and skeletal abnormalities. Homeobox genes make sure your leg isn't where your head should be!
From Wikipedia:
A homeobox is a DNA sequence, around 180 base pairs long, found within genes that are involved in the regulation of patterns of anatomical development (morphogenesis) in animals, fungi, plants, and numerous single cell eukaryotes.[2] Homeobox genes encode homeodomain protein products that are transcription factors sharing a characteristic protein fold structure that binds DNA to regulate expression of target genes.[3][4][2] Homeodomain proteins regulate gene expression and cell differentiation during early embryonic development, thus mutations in homeobox genes can cause developmental disorders.[5]
Homeosis is a term coined by William Bateson to describe the outright replacement of a discrete body part with another body part, e.g. antennapedia—replacement of the antenna on the head of a fruit fly with legs.[6] The "homeo-" prefix in the words "homeobox" and "homeodomain" stems from this mutational phenotype, which is frequently observed when these genes are mutated in animals. The homeobox domain was first identified in a number of Drosophila homeotic and segmentation proteins, but is now known to be well-conserved in many other animals, including vertebrates.[3][7][8]
CAAT and TATA are promoters necessary to start transcription. CAAT is 75 bases upstreat from the start codon and the TATA (Hogness) box is 25 bases upstream from the start codon. The promoters are where RNA pol II and transcription factors bind.
Heteroplasmy is mixture of two types of genetic material; it's the fact that some cells have normal mitochondria and others have mutated mitochondria because during mitosis, the mutation may distribute more to some cells than others. This affects the severity of the disease. Due to random chance, one offspring may be more severely affected than the other because that's just how the mutation distributed amongst cells during mitosis. This is what happens in syndromes such as MELAS syndrome (Mitochondrial Encephalopyopathy with Lactic Acidosis and Stroke-like episodes), which is due to mutation of mtDNA. All the offspring of an affected mother will be affected because you only get mtDNA from your mom, whose eggs have a lot of it. MELAS syndrome causes seizures, stroke-like episodes, muscle weakness, lactic acidosis.
G6PD deficiency is X-linked recessive; this means that affected males will make unaffected sons (because they can't give their sons the bad X) and carrier daughters (who don't have the disease themselves because they have one good X to counteract the bad X). Females who carry X-linked recessive chromosomes have a 50% change of having affected sons and a 50% chance of having carrier daughters.
An enhancer sequence is found in the introns, upstream, or downstream of a gene. In eukaryotes, RNA pol II makes mRNA from DNA template; enhancer sequences bind to activator proteins that help DNA to bend, which lets the activator proteins interact with the transcription factors and RNA pol II, which causes faster transcription. Silencers bind to repressor proteins and decrease rate of transcription.
Hemophilia A is X-linked recessive = factor VIII deficiency.
Nucleosomes = DNA wrapped around a core of 8 histone proteins. Histone H1 is outside of the histone core of nucleosomes and promotes compaction of heterochromatin.
Ok, I think it's prokaryotes that have DNA pol I, II, and III and eukaryotes that have RNA pol I, II, and III. Eukaryotes have 5 DNA polymerases (alpha, beta, gamma, delta, and epsilon). DNA pol I, II, and III have 3'-> 5' exonuclease (proofreading) capability; but only DNA pol I has 5'-> 3' exonuclease activity, which allows it to remove RNA primers and repair damaged DNA. I got more than one question on this. So remember that DNA pol 1 has 5’-> 3’ exonuclease activity. Eukaryotes have multiple origins of replication whereas prokaryotes have 1.
#wobble#bloom syndrome#uniparental disomy#hnRNA#Down syndrome#Robertsonian translocation#mosaicism#genetics#Duchenne muscular dystophy#heteroplasmy#MELAS#MELAS syndrome#G6PD#activator protein#enhancer sequence#friedreich ataxia#Friedreichs ataxia#HOX#hox gene#homeobox#nucleosome#dna polymerase#DNA pol
4 notes
·
View notes
Note
You dont seem like youre fat 🤷♂️ you look really good
Thank you maybe it’s just body dystrophin
1 note
·
View note
Text
Muscular Dystrophy: Can Stem Cell Treatment Help?

Muscular Dystrophy (MD) is a genetic condition that causes the muscles in your body to weaken and degenerate over time. There are several types of Muscular Dystrophies that can occur, like Becker’s Muscular Dystrophy, Limb-Girdle Muscular Dystrophy, etc. Of these, Duchenne Muscular Dystrophy (DMD) is known to be the most fatal one. However, researchers are exploring novel techniques like stem cell treatment to alter the genetic mutations causing the illness and improve the patient’s quality of life.
What Happens in Duchenne Muscular Dystrophy (DMD)?
This condition occurs when there are mutations in the dystrophin gene that code specific muscle proteins. Dystrophin is responsible for the proper functioning of the muscles, and the lack of it can cause the muscle fibres to be in disarray and degenerate. As a result of improper functioning of the muscles, there is progressive and rapid muscle weakness and loss.
The Dystrophin gene found in the X-chromosome affects men, while most women only remain dormant carriers of the gene. Hence, the condition is typically passed on from either parent to the child. However, in some cases, this is due to a new, spontaneous genetic mutation that occurs during pregnancy.
Read this full article here: https://www.cordlifeindia.com/blog/muscular-dystrophy-can-stem-cell-treatment-help/
0 notes
Text
Duchenne muscular dystrophy (DMD) of the most common hereditary disorders
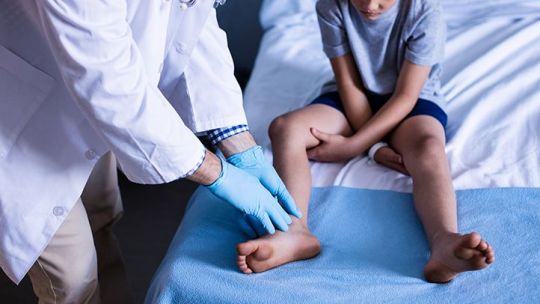
DMD causes muscle weakness when muscle fibres are disrupted, die, and are replaced with connective tissue or fat. The voluntary muscles of the hips, pelvic area, thighs, and calves are the first to be affected. The pain subsequently extends to the shoulders and neck, followed by the arms, respiratory muscles, and other body regions. Fatigue is a common problem. Duchenne muscular dystrophy is one of the most common hereditary disorders, affecting around one in every 3,500 male births throughout the world.
DMD is caused by a mutation in the dystrophin gene at the Xp21 region on the X chromosome's short arm. Dystrophin is a protein complex that connects each muscle fiber's actin cytoskeleton to the underlying basal lamina (extracellular matrix) via a protein complex with numerous components. In the absence of dystrophin, excess calcium can pass through the sarcolemma (the cell membrane). Water enters the mitochondria as a result of changes in calcium and signalling pathways, causing the mitochondria to rupture.
Read more @ https://cmiinfoistic.blogspot.com/2021/10/know-more-about-duchenne-muscular.html
#coherentmarketinsightsreport#coherent market insights#duchenne muscular dystrophy#phramaceutical#muscular dystrophy
0 notes
Last Seen Blogs

td-rarepairs
total rarepair island


blamethecity
The only time you will ever know what I'm thinking

gokyuzugibiissiz
sonsuzlugum

akriloth
Akriloth Space