#fda forms
Text
An In-Depth Guide to Form FDA 1572
What is the Form FDA 1572?
The Form FDA 1572 is a form issued by the United States Food and Drug Administration (FDA). It is a document that must be completed and submitted by sponsors and investigators conducting clinical trials in the United States. This form provides the FDA with important information about the clinical trial, such as the protocol, investigator qualifications, and other important information.
CCRPs provides principal investigator certification to ensure accuracy and efficiency for form completion and compliance.
Who is Required to Complete the Form FDA 1572?
The Form FDA 1572 must be completed and submitted by sponsors and investigators involved in clinical trials conducted in the United States. The sponsor is the organization that is responsible for the conduct of the clinical trial, and the investigator is the individual responsible for the conduct of the trial at a specific site.
How to Complete the Form FDA 1572?
Completing the Form FDA 1572 can be a daunting task for investigators and sponsors. However, it is important to understand the information requested on the form, as well as how to accurately and completely fill out the form. A few tips for completing the form include: thoroughly reviewing the instructions before filling out the form, carefully reading each item on the form and providing complete and accurate information, and providing all required information, such as signatures and dates.
Steps to Filling out FDA 1572 Form:
Write the name of the investigator at the top of the form. For example, enter “John Smith” as the Investigator Name.
Enter the address of the investigator in the next line. For example, enter “123 Main Street, Anytown, USA 12345” as the Investigator Address.
Enter a phone number for contact purposes in either a local or international format (e.g., “1-800-555-1234” or “+1 123 456 7890”).
Enter a valid email address associated with the investigator in the provided field (e.g., [email protected]).
List any previous investigational drug and device studies that have been performed by this investigator under FDA oversight (if applicable). For example, enter “CT-001, DB-002” as Previous Investigational Studies Conducted Under FDA Oversight.
Indicate whether you are requesting approval to conduct clinical trials with drugs or devices by checking one of two boxes: Drugs or Devices/Biologics/Medical Devices/Other Products Regulated by FDA (e.g., select “Drugs” if you are requesting approval for clinical trials with drugs).
Follow up with information about which specific drugs or devices will be used in your studies (e.g., enter “Lipitor, Celebrex” for drugs and/or “Defibrillator XF7500, Pacemaker YZ2300” for medical devices).
Specify how many new indications or dose regimens you will be studying with each drug or device (e.g., enter 2 for Lipitor and 1 for Celebrex).
Provide details about any preclinical studies conducted to evaluate safety and efficacy data related to your proposed clinical trial (if applicable; e.g., provide details about animal models used and results obtained from these tests)
Describe any other research activities related to FDA product regulation that have been conducted by yourself or associates at your organization (if applicable; e.g., enter “Phase II safety study on Lipitor conducted in 2018”)
Sign and date the form after carefully reviewing all information entered into it
FAQs for Form FDA 1572
What is the Statement of Investigator, Form FDA 1572?
The Statement of Investigator, Form FDA 1572, is a document that must be completed and signed by the lead investigator for each clinical investigation conducted under an Investigational New Drug Application (IND). It is used to provide information about the qualifications of investigators conducting studies with investigational drugs.
Why does this form need to be completed by an investigator?
This form needs to be completed by an investigator to ensure that they are qualified and have the necessary experience and expertise to conduct a safe and ethical clinical trial. This form also serves as affirmation from the investigator that he or she has read and understood the protocol of the clinical investigation in question, as well as any other information pertinent to the study provided by the sponsor or sponsor-investigator.
When must this form be completed and signed by an investigator?
The form must be completed and signed by an investigator at or before initiation of a clinical investigation which involves use of an investigational drug. The form must also be updated or a new 1572 must be completed and signed by an investigator if there is new or changed information relevant to the study.
Must the investigator be a physician? What are the minimum qualifications of an investigator?
An investigator does not need to be a physician, but should meet certain criteria set forth by FDA such as having sufficient training, knowledge, and experience pertinent to the type of research being conducted; having access to medical records relevant to studies being conducted; understanding good clinical practice requirements; following protocols; and obtaining informed consent from research participants.
Does the 1572 need to be submitted to FDA?
Yes, this form needs to be submitted to FDA along with supporting documents prior to initiation of a clinical trial involving use of an investigational drug. Even if a foreign clinical study is not conducted under an IND, investigators who conduct such studies still may need to sign a 1572 in certain circumstances.
If a clinical investigation is not conducted under an IND or is for a medical device, must investigators sign a 1572?
A sponsor may conduct a foreign clinical study under an IND only in situations where it does not qualify for exemption from IND regulations due to lack of assurance that subject protection will be maintained without oversight from FDA. If such conditions are met then sponsors must submit an IND application prior initiating the foreign study in order for it to comply with applicable regulations.
Must investigators who conduct studies outside of the United States sign a 1572?
Yes, according to the Food and Drug Administration (FDA), all clinical investigators conducting studies on FDA-regulated products that require an Investigational New Drug (IND) application must sign a Form FDA 1572. This form is used to confirm that the investigator understands their obligations and responsibilities related to conducting IND-related studies.
If a foreign clinical study is being conducted under an IND, what are the investigator's responsibilities with respect to local laws and regulations?
When conducting foreign clinical trials under an IND, investigators must comply with both local laws/regulations as well as those set forth by the FDA in 21 CFR Part 312. This includes ensuring that good clinical practice standards are followed and that any applicable ethical considerations are taken into account when designing and implementing the study protocol. In order to ensure compliance with local laws, investigators may need to obtain permission from national or regional regulatory authorities before beginning the trial. Additionally, depending on the country in which a foreign clinical trial is conducted, additional requirements such as language translations of informed consent forms may be necessary.
For foreign clinical studies conducted under an IND, how can an investigator sign the 1572 when he/she knows he/she cannot commit to all of the requirements on the form, specifically IRB membership (21 CFR 56.107)?
In order for an investigator to sign a Form FDA 1572 for a foreign clinical study under an IND even if they know they cannot commit to all of its requirements (specifically IRB membership), they should discuss this issue with their sponsor prior to signing it in order to find out what alternative arrangements can be made. Furthermore, sponsors should consider both local laws/regulations as well as ICH standards when making these arrangements so that appropriate safety measures can be taken. For instance, sponsors may choose to contract independent consultants or external experts who are familiar with good clinical practice standards in order to review data gathered during trial activities at sites located outside of United States jurisdiction.
If a sponsor chooses to conduct a foreign clinical study (or operate non-US sites in a multinational study) under an IND and the investigators at these non-US sites comply with ICH E6 Good Clinical Practice Consolidated Guidance, would the non-US investigators also be in compliance with FDA's IND requirements under 21 CFR Part 312?
When conducting foreign clinical trials under an IND, compliance with ICH E6 Good Clinical Practice Consolidated Guidance alone may not guarantee full compliance with 21 CFR Part 312 requirements set by the FDA. Although ICH standards provide general guidance on how research should be conducted ethically and safely within different jurisdictions around world, some countries have rules or regulations in place which differs from those established by ICH E6 Good Clinical Practice Consolidated Guidance or which might amend them slightly; therefore potential discrepancies between these two sets of regulations need to be taken into consideration when designing trial protocols for international trials subject to FDA jurisdiction. Furthermore, sponsors should ensure that all parties involved in such trials understand their individual responsibilities related executing Research Ethics Committee approval processes required for each country included in study protocol design prior commencing trial activities at each site outside US jurisdiction
Must foreign clinical study sites in a multinational study that includes domestic sites be conducted under an IND?
Yes, all foreign clinical study sites that are part of a multinational study must be conducted under an IND. The sponsor must submit an application to the FDA for approval to conduct the study and provide detailed information about the site, such as personnel qualifications, resources and facilities available at the site, and protocol for conducting the research. The IND application includes protocols and other information describing how a proposed clinical investigation will be conducted.
How does a sponsor submit information to FDA about a foreign clinical study that was not conducted under an IND?
The sponsor must submit an Investigational New Drug (IND) Application to the FDA if they wish to conduct a foreign clinical study which has not been previously approved by the FDA. The sponsor should include detailed information regarding the proposed clinical trial, including the proposed protocol, safety measures put in place to protect subjects participating in the trial, qualifications of personnel involved in conducting or supervising the trial, and any other information which will help demonstrate compliance with applicable regulations.
Should a new form be prepared and signed when the OMB expiration date is reached?
No, there is no need for sponsors to prepare or sign any new forms when submitting an Investigational New Drug (IND) Application or when seeking approval from FDA for any particular clinical trial. However, sponsors must follow all applicable laws and regulations related to their research activities and comply with requirements set forth in relevant documents such as Form 1572 (Declaration for Clinical Investigations Involving Human Subjects), Form 3454 (Statement of Investigator), and Form 3753A (Clinical Investigator's Brochure).
Does FDA expect a double-sided 1572, or is a two-page document printed from the FDA website acceptable?
The FDA requires sponsors to submit Form 1572 as part of their IND application as both single-sided copies and double-sided copies. The form should be completed according to applicable regulations outlined by 21 CFR 312.23(a)(7). Sponsors may not use double-sided copies of documents obtained from websites hosted by other organizations, including those belonging to different government agencies or non-profit institutions..
How should the 1572 be completed?
Form 1572 should be filled out completely by each investigator listed on it who is responsible for conducting or supervising certain aspects of research activities at any given site. This includes providing all necessary details such as person’s name, address/location(s), contact information (e-mail address/phone number/fax number etc.), signature(s) etc., along with listing any degrees/licenses held by him/her that show he/she is qualified to conduct/oversee said research activities being funded through this particular project. Furthermore important section detailing ‘Financial Disclosure’ needs special attention especially since this form also serves purpose of informing potential participants about potential conflicts of interest pertaining to investigator’s involvement in these studies alongside his/her salary details etc. So it is crucial that this section is filled out completely without leaving out any significant details so that true picture can be presented in front of future volunteers who might decide whether they want participate in said studies or not based on aforementioned disclosure
Review Questions for FDA Form 1572
What is FDA Form 1572?
A) A form that must be completed and signed by the clinical investigator when a study is initiated, revised, or discontinued
B) A form that must be completed by all patients participating in a study
C) A document used to report adverse drug events to the FDA
D) A document used to collect information about the safety and effectiveness of drugs
Answer: A) A form that must be completed and signed by the clinical investigator when a study is initiated, revised, or discontinued. Explanation: The FDA Form 1572 is an agreement between investigational sites and the FDA. It outlines key elements of studies conducted at those sites such as background qualifications of investigators and staff, source documents, records maintenance, reporting requirements and procedures for handling drugs used in clinical trials.
What type of information must be provided when completing FDA Form 1572?
A) Personal information about each individual participant in a trial
B) Information about drugs being tested in a trial
C) Financial information from sponsors involved in the trial
D) Information about laboratory tests performed during the trial
Answer: B) Information about drugs being tested in a trial. Explanation: The FDA Form 1572 requires that the investigator identify all drugs to be administered during the investigation (e.g., active ingredient names and doses), along with any other products that may affect laboratory results such as vitamins or minerals. This will help ensure accurate record keeping throughout the trial.
Who is responsible for ensuring accuracy on FDA Form 1572?
A) The clinical investigator conducting the study
B) The sponsor of the study/trial
C) The patient participating in the study/trial
D) All of the above
Answer: D). All of the Above. Explanation: Accuracy on FDA Form 1572 is essential since it serves as an agreement between investigational sites and the Food & Drug Administration (FDA). Thus, both sponsors and clinical investigators are responsible for ensuring accuracy on this form, as well as patients who participate in studies/trials should they provide any data or information required by this form.
When does an individual need to submit an updated version of FDA Form 1572?
A) When enrolling new patients into a clinical trial
B) When changes are made to protocols related to a given clinical trial
C ) When making changes to personnel associated with a given clinical trial
D ) All of the above
Answer: D). All of the Above Explanation: An updated version of FDA Form 1572 needs to be submitted when enrolling new patients into a given trial; when changes are made to protocols related; or when personnel associated with a given clinical trail have changed since its initiation or last update. This helps ensure accuracy so that all parties involved have access up-to-date information regarding ongoing studies/trials they’re involved with at any given time.
What happens if an individual fails to submit an updated version of FDA Form 1572?
A ) They will not receive funding for their research project
B ) Their research project may not pass inspection from regulatory authorities
C ) They may face legal repercussions from regulatory authorities
D ) All of the Above
Answer: D). All of The Above Explanation: If an individual fails to submit an updated version of FDA Form 1572 then they can face various consequences such as not receiving necessary funding for their research project; having their research project fail inspection upon review by regulatory authorities; or facing legal repercussions from said authorities due its importance in providing complete documentation related to ongoing studies/trials involving human subjects which helps protect participants’ rights while conducting necessary research work safely and ethically within regulatory guidelines set forth by law enforcement bodies responsible for protecting public health around world according these standards set forth through years long process establishing best practices medical community has come accept today across many countries globally depending respective jurisdiction laws apply under question particular case being consider review possible action taken based findings presented within scope parameters policy established maintain highest ethical standard ensure well-being everyone involved
CCRPs provides principal investigator certification to ensure accuracy and efficiency for form completion and compliance.
#fda full name#fda form 1572#1572 form fda#match the following statements with the appropriate tissue sample.#1572a#fda form 3455#clinical forms#fda form#conducting investigator-initiated studies according to fda regulations and gcp#fda form 3674#1572 fda guidance#fda guidance on 1572#1572 template#form 1572 fda#1572#fda forms#conducting investigator initiated studies according to fda regulations and gcp#fda 3455 form#form fda 3454#fda investigator#nursing task#fda 1572 statement of investigator#fda clinical trial#1572 form#1572 fda#fda 1572 guidance#dd quiz#when must the investigator update the irb#fda form 3454#fda1572
0 notes
Text
A hopeful time for Cryptosporidium research - Technology Org
New Post has been published on https://thedigitalinsider.com/a-hopeful-time-for-cryptosporidium-research-technology-org/
A hopeful time for Cryptosporidium research - Technology Org
Due to the many technical difficulties studying Cryptosporidium, scientists have struggled for many years to advance research on the single-celled parasite that is one of the leading causes of deadly diarrheal disease. Multiple breakthroughs in the past decade, says biologist Boris Striepen of the School of Veterinary Medicine, have made this a tractable pathogen and disease.
A lot of research progress has been over the past decade on
Cryptosporidium, a single-celled parasite that is one of the leading causes of deadly diarrheal disease, and Penn Vet professors brought together researchers and clinicians from around the world for a conference. Image credit: Muthgapatti Kandasamy and Boris Striepen
With support from the Bill & Melinda Gates Foundation and the National Institutes of Health, the Striepen Lab and others have pursued an ambitious research agenda. Genetic engineering of the parasite and new culture and animal models enabled progress toward drugs and vaccines. New candidate drugs have entered human trials for the first time in many years.
Striepen and Christopher Hunter, also of Penn Vet, sought to amplify these advances by organizing the First Biennial Cryptosporidium Meeting, held at Penn. It included academic researchers from across disciplines, scientists from leading pharmaceutical companies, representatives of United States and international public health agencies, and leading clinicians from some of most impacted countries, including Zambia, Kenya, Colombia, Bangladesh, and India.
“There had been transformational progress, and we thought this a great opportunity to bring everybody together to ask. Now that we have the tools to address this problem, where is the field and what should we do next?” Striepen says.
He stresses the gravity of cryptosporidiosis—the disease caused by the parasite—and the importance of finding drugs and vaccines. The conference spanned the field, from the fundamental biology of the life cycle of Cryptosporidium to the state of drug development and challenges of clinical trials for the disease, which is most prevalent in highly vulnerable babies and toddlers.
Striepen says 10% of child mortality worldwide comes from diarrheal disease, and, after rotavirus, Cryptosporidium is a main cause. The disease tracks with poverty, and low-income regions are most affected. “It has this vicious cycle relationship with malnutrition, so malnourished kids are very susceptible,” he says, “but having this infection also sets kids up for future malnutrition.”
Cryptosporidiosis, says Striepen, was not appreciated as a human disease until it was identified as an AIDS-defining illness in the 1980s. Increased attention and improved diagnostics showed that others frequently suffer from cryptosporidiosis but recover without treatment if they have healthy immune systems. Striepen says that half of U.S. disease outbreaks linked to recreational water are due to this parasite because its infectious stage is resistant to water chlorination.
Striepen is hopeful not only because of how much progress has been made on Cryptosporidium but also because of how many young scientists and physicians attended the conference. Two such people are fifth-year immunology Ph.D. student Breanne Haskins and postdoctoral fellow Aurelia Balestra, who both came to Penn specifically to work on Cryptosporidium.
Haskins works on the T cell response to the parasite, which she says is important because people who lack T cell responses can remain chronically infected or suffer from repeat infections. Haskins adds that the lone FDA-approved drug is not effective in some individuals, such as those with HIV/AIDS. Specifically, Haskins researches the components from the parasite and host that are necessary to induce T cells, which she says could potentially identify future vaccine targets.
Along with the negative impact Cryptosporidium infection has on children and the need for a vaccine, one of Haskins’ takeaways from the conference was that many cases go undiagnosed because diagnostic tools aren’t accessible in lower-income countries. She says that “we need better diagnostics in order to know individuals are infected with Cryptosporidium if we want to administer an effective drug.”
Balestra adds that cryptosporidiosis is not on the World Health Organization’s list of neglected tropical diseases despite meeting the criteria. She argues that including it is critical to increasing awareness, securing funding, and improving disease monitoring.
In a session with other molecular biologists at the conference, Balestra gave a talk about sexual development and fertilization in Cryptosporidium. She explains that, just as humans have sperm and eggs, Cryptosporidium produces male and female gametes. However, these parasite gametes differ significantly from human ones, so scientists don’t know how the male and female gametes fuse to form a zygote. Her research focuses on finding out, and she has demonstrated that sexual development is essential for the parasite’s growth.
Balestra says it is an exciting time to work on this parasite because of the availability of new tools and that people at the conference agreed that “the increasing amount of people working on tackling this disease gave hope to be able to treat the patients in the future better.”
Source: University of Pennsylvania
You can offer your link to a page which is relevant to the topic of this post.
#1980s#amp#attention#awareness#Babies#Biology#Biotechnology news#cell#Cells#Children#Companies#conference#cryptosporidium#development#diagnostics#diarrhea#Disease#Diseases#drug#drug development#drugs#engineering#FDA#Fertilization#form#Foundation#Fundamental#Funding#Future#genetic
0 notes
Photo

FDA Form 483 response in USA:- If you are searching for an international food safety consulting and testing firm, then look no further than us. We help with GFSI consulting and also provide food safety consulting. Call us to collect further details. for more info visit us: https://ghpconsultingandtesting.com/
0 notes
Text

#According to the manufacturer#Claritox Pro is a product formed completely of natural components produced by organic planters. They have also paid close attention to usin#This Claritox Pro supplement is manufactured in a US facility. Therefore#it is not only GMP-certified#FDA-approved#sterile#and safe but also does not use non-GMO ingredients#stimulants#and impurities.#The Claritox Pro formula was developed by Jim Benson#a 67-year-old Tennessee researcher. What is most significant to him is plants and their effectiveness in boosting the soundness and functio#https://www.claritoxproo.com/ctp-tsl
0 notes
Link
Are you searching for the best food safety consulting companies? If so, look no further than GHP Consulting and Testing LLC. We also help with chemical testing, microbiological testing, calibration of equipment, etc. See it here. https://ghpconsultingandtesting.com/service/general-food-safety-quality-support/

0 notes
Link
Are you searching for the best food safety consulting companies? If so, look no further than GHP Consulting and Testing LLC. We also help with chemical testing, microbiological testing, calibration of equipment, etc. See it here. https://ghpconsultingandtesting.com/fssc-22000-consultant

0 notes
Text
Histotripsy works by using targeted ultrasound waves to form microbubbles within the tumor. The forces created as those bubbles form and collapse cause the mass to break apart, killing tumor cells and leaving the debris to be cleaned up by the immune system.
What that could mean for patients is treatment without the physical toll of radiation or chemotherapy, fewer concerns with drug compatibility, far shorter recovery times than with surgery and less treatment discomfort.
And histotripsy’s potential benefits go beyond tumor destruction. In the last year, a pair of pre-clinical studies in rodents suggest that in the clean-up process, the immune system learns how to identify cancer cells as threats. This can enable the body to continue fighting the initial tumor and help activate a natural immune response to the cancer.
In the first study, even after destroying only 50% to 75% of the liver tumor volume by histotripsy, the rats’ immune systems were able to clear away the rest, with no evidence of recurrence or metastases in more than 80% of animals.
1K notes
·
View notes
Text
6 Tips to avoid 483 letters
The U.S. Food and Drug Administration (FDA) is authorized to perform random inspections and audits, and these inspections can lead to FDA warning letters and FDA Form 483 observations.
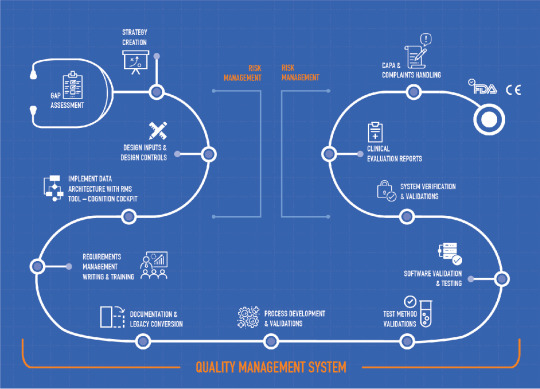
Medical device manufacturers receive observations (Form 483) and / or warning letters on completion of USFDA Audit. The document outlines any violations of Good Manufacturing Practices (GMP’s) such as the facility, equipment, processes, controls, products, employee practices or records.
Some tips to avoid a warning letter and form 483 observation –
· Be Inspection Ready
· Clearly Written Standard Operating Procedures
· Proper Document Control
· Develop a Compliance Culture
· Correct Observations Real-time
· A Hyper-adaptive Quality Management Software
IZiel has successfully assisted medical device companies to resolve these observations with efficiency and accuracy. IZiel team in collaboration with the customer utilize an extensive & comprehensive methodology to complete the remediation project. Our Onshore-Offshore Model with strong project management ensure deliveries with faster timelines, flexible resources and in cost-effective manner.
0 notes
Text
The Best News of Last Week - March 18
1. FDA to Finally Outlaw Soda Ingredient Prohibited Around The World

An ingredient once commonly used in citrus-flavored sodas to keep the tangy taste mixed thoroughly through the beverage could finally be banned for good across the US. BVO, or brominated vegetable oil, is already banned in many countries, including India, Japan, and nations of the European Union, and was outlawed in the state of California in October 2022.
2. AI makes breakthrough discovery in battle to cure prostate cancer
Scientists have used AI to reveal a new form of aggressive prostate cancer which could revolutionise how the disease is diagnosed and treated.
A Cancer Research UK-funded study found prostate cancer, which affects one in eight men in their lifetime, includes two subtypes. It is hoped the findings could save thousands of lives in future and revolutionise how the cancer is diagnosed and treated.
3. “Inverse vaccine” shows potential to treat multiple sclerosis and other autoimmune diseases
A new type of vaccine developed by researchers at the University of Chicago’s Pritzker School of Molecular Engineering (PME) has shown in the lab setting that it can completely reverse autoimmune diseases like multiple sclerosis and type 1 diabetes — all without shutting down the rest of the immune system.
4. Paris 2024 Olympics makes history with unprecedented full gender parity

In a historic move, the International Olympic Committee (IOC) has distributed equal quotas for female and male athletes for the upcoming Olympic Games in Paris 2024. It is the first time The Olympics will have full gender parity and is a significant milestone in the pursuit of equal representation and opportunities for women in sports.
Biased media coverage lead girls and boys to abandon sports.
5. Restored coral reefs can grow as fast as healthy reefs in just 4 years, new research shows

Planting new coral in degraded reefs can lead to rapid recovery – with restored reefs growing as fast as healthy reefs after just four years. Researchers studied these reefs to assess whether coral restoration can bring back the important ecosystem functions of a healthy reef.
“The speed of recovery we saw is incredible,” said lead author Dr Ines Lange, from the University of Exeter.
6. EU regulators pass the planet's first sweeping AI regulations

The EU is banning practices that it believes will threaten citizens' rights. "Biometric categorization systems based on sensitive characteristics" will be outlawed, as will the "untargeted scraping" of images of faces from CCTV footage and the web to create facial recognition databases.
Other applications that will be banned include social scoring; emotion recognition in schools and workplaces; and "AI that manipulates human behavior or exploits people’s vulnerabilities."
7. Global child deaths reach historic low in 2022 – UN report

The number of children who died before their fifth birthday has reached a historic low, dropping to 4.9 million in 2022.
The report reveals that more children are surviving today than ever before, with the global under-5 mortality rate declining by 51 per cent since 2000.
---
That's it for this week :)
This newsletter will always be free. If you liked this post you can support me with a small kofi donation here:
Buy me a coffee ❤️
Also don’t forget to reblog this post with your friends.
734 notes
·
View notes
Text
Research Seeks to Break the Mold of Ultra-Lightweight Aerogels - Technology Org
New Post has been published on https://thedigitalinsider.com/research-seeks-to-break-the-mold-of-ultra-lightweight-aerogels-technology-org/
Research Seeks to Break the Mold of Ultra-Lightweight Aerogels - Technology Org
Producing ultra-lightweight materials that are also strong could revolutionize multiple industries. With groundbreaking work from an interdisciplinary team of chemists and 3D printing experts, that revolution could be closer.
Garrett Godshall inspects a 3D-printed piece of aerogel produced in the partnership between the labs of Robert Moore and Chris Williams. Illustration by Alex Parrish for Virginia Tech.
Aerogels are a unique class of ultra-low-density materials with a weight only about 15 times heavier than air. If an average adult were made of aerogel, they would weigh somewhere between 3 and 14 pounds.
The material has been around for a little less than 100 years, and the first aerogels were a highly porous solid containing more than 99 percent air, nicknamed “frozen smoke.” Although these aerogels made from silica glass have achieved the Guinness World Record for the lowest density solids, they are known to be very brittle and quite expensive to process.
Today, the race is on to find new materials and cost-effective methods to produce incredibly strong aerogels for advanced applications such as thermal insulation for aerospace vehicles, passive solar insulation for next generation housing, water and air filtration, lightweight packaging, controlled drug delivery, and personally tailored biomedical scaffolding.
That race has some strong Virginia Tech contenders. Robert Moore, a professor in the Department of Chemistry in the College of Science, has joined forces with Christopher Williams, the LS Randolph Professor of Mechanical Engineering in the College of Engineering. They have brought together the resources of both their groups to produce new approaches to engineered aerogels, channeled through the Macromolecules Innovation Institute. Graduate researchers Garrett F. Godshall and Daniel A. Rau have led those teams with innovations in both materials and machinery with the methods published in the journal Advanced Materials.
An infrared camera image of polyphenylene sulfide gel during the 3D printing process. Illustration by Robert Moore, Virginia Tech.
Strong, lightweight skeletons
The first step to making an aerogel involves producing a gel, a 3D solid network that entraps a liquid, like water in gelatin. The next step involves carefully removing the liquid in the gel, leaving behind an ultralight microporous sponge-like skeleton because the heavy liquid has been replaced by air and is potentially strong because of its interconnected 3D lattice.
One material that had not previously been developed as an aerogel is polyphenylene sulfide (PPS), a super strong thermoplastic often used as a substitute for metal when weight reduction and chemical resistance is required. As an aerogel, PPS could usher in a new wave of applications, particularly in lightweight high performance thermal insulation. Recently, Moore’s research group has demonstrated a simple process for creating PPS gels and aerogels using a unique nontoxic, environmentally friendly solvent.
Moore’s team brought its process to Williams, who has pioneered novel 3D printing methods.
“Bob and I have been working together for many years thanks to the interdisciplinarity fostered through the Macromolecules Innovation Institute,” said Williams. “We hosted a joint meeting between our two groups over a summer so that our students could become more aware of our labs’ capabilities and expertise and ideate ways we could collaborate. From that meeting, we identified that Bob’s novel approach for synthesizing PPS aerogels would meld well with my group’s expertise in 3D printing and prior work in printing PPS.”
“Unlike the silica aerogels or other crosslinked polymers used by NASA, our PPS gels don’t require complex chemical reactions and they can be melted and solidified over and over again,” said Moore. “All we have to do is make a hot solution of commercially available PPS and then cool it to room temperature. Using our new, safe solvent, which is actually an FDA-approved food grade additive, the PPS solutions gel in seconds. It is as simple as making Jell-O. But once we saw the super-fast solidification of these gels, we knew it was time to team up with the Williams group to see if we could print this stuff.”
The PPS aerogel barrier has been breached with Moore’s discovery of rapid PPS gelation. Using a combination of simple chemistry and 3D printing innovation, the first additive manufacturing of PPS into an aerogel is now a reality.
Breaking the mold
Like gelatin, the gels that are used to make aerogels are conventionally formed in open molds. This yields a solid form with limited size and shape. Making PPS aerogels with engineered shapes and geometries requires a combination of innovations in polymer chemistry and advanced manufacturing.
On Moore’s team, Godshall produced pellets of the PPS gel and placed them into a new, high temperature printing tool designed by Rau specifically for this task. Inside the nozzle, the gel pellets are re-liquified and extruded onto a substrate, where they cool and re-solidify.
After the print is finished, the solvent-containing gel part has the solvent removed through an exchange process and freeze drying, resulting in a PPS aerogel. This process enables the formation of microscopic pores that can be tuned by the print settings. Moreover, at the macroscale, the three-dimensional form of the PPS aerogel can be tailored by the infinite shape possibilities of 3D printing.
Creating large, lightweight shapes to the contour of an airplane wing or small insulating structures incorporated in electronic devices means a reduction of materials in manufacturing; strong aerogel frameworks could equate to a reduction of fuel with lighter vehicles, and engineered thermal insulators could advance energy efficiency in next-generation technologies.
“This publication represents a significant breakthrough in the manufacturing of complex aerogels from engineering polymers,” said Williams. “Bob’s synthesis technique can work for a number of other high-performance polymers, and the printing process can be easily modified to account for these changes. We were surprised to learn that the processing conditions have an effect over the morphology and density of the aerogel structure. We are excited to study this further and discover how to gain control and program this structure and performance into new multi-functional parts.”
Source: VirginiaTech
You can offer your link to a page which is relevant to the topic of this post.
#3d#3D printing#additive manufacturing#advanced materials#aerogel#aerogels#aerospace#air#applications#approach#barrier#chemical#chemical reactions#chemistry#Chemistry & materials science news#collaborate#college#devices#drug#drug delivery#efficiency#electronic#electronic devices#energy#energy efficiency#engineering#FDA#Food#form#freeze
0 notes
Text
Would Batman Rogues Tamper With Halloween Candy?
Joker: Plants just enough evidence to make it look like maybe 1 out of 100 pieces of candy will have Joker toxin in them, watches Gotham whip itself into a frenzy and deny itself and its children their Halloween fun, then hijacks the news the next morning to reveal that he hadn't spiked anything at all--the reveal ends with a play on the two meanings of "sucker".
Riddler: The stubborn pervasiveness of such a blatant falsehood annoys him so much that he refuses to dignify it by making it true.
Catowman: That is simply not her M.O.
Penguin: Representative of the "Drugs cost money, why would I hand them out to people with no money who won't be able to identify or trace what they got high on?" camp. That said, he might squeeze in on a technicality, because any candy manufacturer that he owns is definitely breaking all the FDA regulations.
Poison Ivy: Maybe dosing the children of high-level corporate execs with the illnesses their waste production sets loose on third-world countries? But more likely she's shoring up her resources to combat the Yearly Evergreen Massacre.
Mr. Freeze: Would this advance research into curing Nora's illness? Would it bring in money he could use to keep the cryo-chamber running? Then why would he do it?
Mad Hatter: Yes, absolutely, no question about it. But not for any of the more sordid reasons that one might suspect. No, this is a showcase for his skill with hallucinogens, rather than outright control, as well as his misapplied sense of whimsey. The very city of Gotham itself is going as Wonderland for Halloween...whether it likes it or not.
Scarecrow: Contrives to lace every single piece of candy in the city with a highly diluted form of fear toxin, that raises your pulse and tickles the back of your neck and makes the shadows looks a little deeper, in the name of fostering the "Halloween spirit".
#batman#batman rogues#Two-Face isn't on here because his entry would just have been a repeat of Catwoman's
303 notes
·
View notes
Text
People who get all up in arms about vaccination programs in the US because "it's un-American for the government to be involved in my healthcare" are just a special level of stupid.
I mean first of all government vaccine programs have been going on for decades, it's how we almost eradicated polio from the country and significantly brought down the rates of measles, not to mention y'know eradicating smallpox thanks to global vaccine cooperation.
But like, people don't realize just how many things they take for granted are the result of government intervention in healthcare. Obviously basic things like working sewage systems, sanitation departments, and the FDA are the first things that come up, but also....
Fluorinated tap water to make sure kids' teeth enamel forms completely
Iodized salt to reduce thyroid issues as a result of iodine deficiency
Fortified cereal, milk, and orange juice to combat vitamin and mineral deficiency
Don't have to worry about scurvy or iodine deficiency? Thank the government intervening in healthcare.
Don't have to worry about cholera, dysentery, or typhus? Thank the government intervening in healthcare.
Don't have to worry about wood shavings and chalk dust in your bread? Thank the government for intervening in healthcare.
The government has always been involved in your healthcare, you've just been taking it for granted.
206 notes
·
View notes
Note
I don't know if this would be useful information for you or any other transmasc folks, but it was news to me until my doctor suggested it: there is an approved pill version of testosterone! It was prescribed to me as part of my gender-affirming care!
https://www.fda.gov/news-events/press-announcements/fda-approves-new-oral-testosterone-capsule-treatment-men-certain-forms-hypogonadism
I'm not sure how widely available it is or which insurance companies will cover it, but it might be good for folks to know it's an option if they're like me and cannot handle needles!
Heya, this is really useful info and I hope the pill form goes smoothly for you!
It behooves all of us on HRT to keep on top of the various ways in which we can get our meds, in case we need to pivot to a different form factor or provider.
Thanks so much for sharing, as this was new to me.
64 notes
·
View notes
Text
Herbalism 101: Lavender

Fun Fact: Lavender is a part of the mint family; making it a natural insect repellent.
English lavender (Lavandula angustifolia) is an excellent way to add a mid-summer pop of color to your garden or patio. Planting is somewhere where you can enjoy its beautiful scent. English lavender is more tolerant of colder temperatures common in the Marquette area than French lavender, making it an excellent choice for herb gardens and perennial borders. This is a member of the mint family features aromatic leaves and flowers that are popular for cooking purposes, medicinal use, and home projects.
Lavender is prized as a culinary ingredient. Lemon and lavender make a great combination for baking cookies, cakes, and other delicacies. Lavender can also be used to infuse simple syrups for cocktails or lemonades.
Lavender’s lovely scent is known to reduce anxiety and calm the mind. The calming fragrance of a lavender plant is thought to provide relief from stress, depression, and migraines. It is also considered anti-inflammatory and has antiseptic properties.
Lavender attracts beneficial pollinators to its flowers. Adding lavender to your garden will attract butterflies, bees, and other helpful insects.
Lavender plants are deer resistant. Making them a welcome addition for many homeowners looking for plants they don’t have to share with the local wildlife.
Lavender from your garden can be dried and used to create items such as homemade lavender soap, bath salts, potpourri, sachets, and cleaning products.
Lavender does well in most climates. The plant is drought resistant and will form a large swath of flowering plants. The blue flowers are not only beautiful but smell fabulous. You can harvest the blooms through the summer and prune the plant back in the fall.
-flower works blog
Lavender is a flowering plant in the mint family that’s easily identified by its sweet floral scent. It’s believed to be native to the Mediterranean, The Middle East, and India, with a history dating as far back as 2,500 years.
According to everyday health blog, Possible health benefits of Lavender include:
May help improve sleep.
Can help treat skein blemishes.
May offer a natural remedy for pain.
Reduce blood pressure and heart rate.
Could relieve asthma symptoms.
Help combat fungus growth.
Potentially promotes hair growth.
Helps relieve stress.
Lavender hasn’t been approved by the FDA, so it’s important to be aware of potential health risks or side effects of using this herb.
📷© Pinterest
#witch#kitchen witch#pagan#witch blog#herbs#witchcraft#kitchen witchery#green witch#hearth witch#herbal magick#witchy#pagan witch#witch aesthetic#witches#witchyvibes#baby witch#cottage witch#earth witch#forest witch#hedge witch#kitchen witchcraft#lunar witch#nature witch#witch community#witch core#witch familiars#witch history#witch tips#witchblr#witchcore
334 notes
·
View notes
Text
Monopolizing turds

Update 31 May 2023: an earlier edition of this article identified the price of Rebyota as $20,000; this was the rumored price prior to Rebyota’s release in December 2022, when Stephen Skolnick wrote the article I referenced. When Rebyota was actually released in 2023, the average wholesale price (AWP) was $10,800. Thanks to Benjamin Jolley for catching this error, and to Stephen Skolnick for getting to the bottom of it.
It’s been ten years — to the day! — since I first started writing about the bizarre, amazing world of turd transplants, in which a sick person receives a microbiotic infusion in the form of some processed poop from a healthy person:
https://web.archive.org/web/20130608030455/http://blogs.plos.org/publichealth/2013/05/29/why-diy-fecal-transplants-are-a-thing-and-the-fda-is-only-part-of-the-reason/
Gut biomes are one of those understudied, poorly understood medical areas that are both very promising and also full of sketchy medical claims from “supplement” companies, influencers, quacks and grifters. But in the decade since I first started tracking turd transplants (formally called “Fecal Microbiota Transplants” or FMTs), a growing body of sound science has emerged on the subject.
One thing that’s increasingly undeniable is that the composition of your microbial nation is related in significant ways to both your physical and mental health. What’s more, as antibiotic resistant “super bugs” proliferate, FMTs are becoming increasingly central to treating dangerous gut infections that otherwise stand a high chance of killing you.
“Eat Shit and Prosper” is Stephen Skolnick’s delightfully named newsletter about poop and health science. Skolnick is a physicist by training, but has a long history of collaboration with Openbiome, a nonprofit that coordinates between doctors, patients and donors to provide safe FMTs:
https://stephenskolnick.substack.com/
In an edition of Eat Shit from last December, Skolnick recounts the amazing history and dismaying future of FMTs. In 2013, the FDA announced it would regulate FMTs as “Investigational New Drugs,” which could only be administered as part of a registered clinical trial:
https://stephenskolnick.substack.com/p/a-monopoly-on-poop
At that point, FMTs were already in widespread use by docs to treat otherwise untreatable cases of Clostridioides difficile (C. diff), an antibiotic resistant bacterial infection that literally makes you shit yourself to death. These doctors were in no position to run registered clinical trials, which meant that they would have to stop using the most effective therapy they had for a potentially lethal infection.
Doctors and patients kicked up a fuss, and the FDA walked back its guidance, announcing that it would exercise “discretion” in enforcing its Investigational New Drug rule, giving a pass to docs who were treating C. diff with FMTs:
https://www.federalregister.gov/documents/2013/07/18/2013-17223/guidance-for-industry-enforcement-policy-regarding-investigational-new-drug-requirements-for-use-of
That’s where things have stood for the past decade or so. The “discretion” rule means that patients could still get FMTs, but their insurance wouldn’t cover it. But even if you had cash to pay for an FMT, your doc probably wouldn’t administer it for anything except a C. diff infection, despite the promising signs that FMT can help treat other conditions, and despite the generally safe nature of FMTs.
If your doc did give you an FMT, chances are good that they sourced their poop from Openbiome. Openbiome recruits very healthy people, gets them to poop in a bag, then processes the poop — removing nonbacterial solids, testing it for pathogens, freezing it, portioning it, and sending it to docs. All this is done at cost, and it’s not cheap: $1–2k/treatment, mostly due to cold-chain logistics (the poop is shipped at -80C).
Despite the cost, and despite the limitations on treatment, the Openbiome method has proved very reliable. Indeed, FMTs as a whole are pretty darned safe, with the most common side-effects being transient gas and bloating. In the past decade, there’ve been a total of six “adverse effects” associated with Openbiome’s 5,000+ procedures, all in severely immunocompromised people, and none conclusively linked to the treatment:
https://www.sciencedirect.com/science/article/pii/S0016508522003511/pdf
A decade into this system, the FDA has taken the next step forward — only it’s actually a step backwards.
During this intervening decade, a pharma company called Ferring has conducted clinical trials on FMTs and received approval for an FMT product called Rebyota. The process for making Rebyota is effectively identical to the process used by Openbiome: collect poop, remove solids, test for pathogens, add glycerol, freeze and ship.
The main difference between Rebyota and Openbiome’s poop is price. While Openbiome charges $1–2k per treatment, Rebyota charges $10,800
That’s some expensive shit!
Fine. Getting Rebyota through clinical trials means that insurers might start covering it, and perhaps some patients will prefer brand-name poop to open-source poop. But as part of the FDA’s approval of Rebyota, the agency also rescinded its “discretionary enforcement” guidance, making it illegal for docs to source their poop from Openbiome:
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/enforcement-policy-regarding-investigational-new-drug-requirements-use-fecal-microbiota
For Ferring, this is a monopoly on shit, one that lets them charge patients $10.8k for poop that costs $1–2k to process. The FDA does not claim that this is being done in the name of safety. Instead, an FDA official told Skonick that the goal was to “incentivize innovation without creating an access crisis.”
That is, the FDA changed its guidance and put nonprofit stool banks out of business because it wants to incentivize pharma companies to perform expensive clinical trials, and it believes that these companies won’t pay for trials if they have to compete with the likes of Openbiome, which would make it impossible to charge 900% markups on poop.
Trials are important! Evidence-based medicine is important! But Ferring’s clinical trials didn’t tell us anything we didn’t already know. FMTs were already the best therapy we had for C. diff. Testing Rebyota against a placebo didn’t tell us anything new — unlike testing Rebyota against the existing therapies, e.g. product from open stool banks.
Such a trial might have given rise to a very different regulatory outcome, because the cure rate reported by Rebyota is much lower than the cure rate from Openbiome’s own interventions:
https://link.springer.com/article/10.1007/s40265-022-01797-x
That is, using the $1k poop from Openbiome seems to be much more effective than using the $10.8k poop from Ferring. But Openbiome, a nonprofit, hasn’t been able to perform the kind of rigorous — and expensive — clinical trial that Ferring funded.
This points to a significant problem with the FDA’s model. The agency wants good clinical data for the medicines it regulates, as it should, It presumes that the only way to get that data is through granting commercial exclusivity to a for-profit, which ends up costing patients vast sums, and locking many patients out altogether.
This creates all kinds of new dangers. 150,000 people/year in the US contract Recurrent Clostridium difficile Infection (RCdI). FMT increases the cure rate by 20% relative to antibiotics alone. That means that if everyone with RCdI gets a poop transplant, 30,000 extra people will get better. That’s a big number!
For well insured people, Rebyota probably represents a cash-savings — if your insurance covers the $10,800 procedure, you might pay $500 out of pocket, which is far less than the $1–2K you’d pay to get an Openbiome poop transplant. But if you’re uninsured or underinsured, the FDA’s new enforcement rules mean that you’re now on the hook for $10,800.
The FDA did carve out a loophole: if your doc or their hospital are willing to prepare the poop transplant themselves, they can administer that. On the one hand, preparing a poop transplant isn’t that hard — some people do them at home, on their own:
https://web.archive.org/web/20211015060558/https://thepowerofpoop.com/epatients/fecal-transplant-instructions/
But on the other hand, there’s been exactly one death conclusively linked to FMT, and it was from one of these hospital-prepared transplants (the patient had just had a marrow transplant for cancer that wiped out their immune system, and the donor had a novel pathogen that the hospital failed to test for).
So the FDA has created a situation where, if you can’t afford a $10,800 proprietary formulation, your only option is to convince your doc or hospital to prepare their own poop transplant, which will cost less than the $10.8k for Rebyota, but more than the $1–2k from Openbiome, which has all kinds of economies of scale. And if you do manage it, you’ll be getting a procedure that has a much worse safety track-record than the Openbiome process that the FDA just killed.
The FDA has an important role to play here, but as with so many policy questions, how the FDA plays that role depends on things that are far upstream from the agency and its decisions. The choice to fund medical trials through the promise of exclusivity — and with it, extremely high margins — puts the FDA in the position of choosing winners in the marketplace: Ferring wins, Openbiome loses.
Ironically, this is the thing that exclusivity is supposed to prevent. By using profit to incentivize medical research, the FDA is supposed to be recruiting the Invisible Hand as its partner in regulation. But exclusivity is incompatible with the idea of medicine as a public good. The tens (hundreds) of millions that Americans will pay for $10.8k poop transplants from Ferring will add up to far more than it would cost to underwrite clinical trials for an open process like Openbiome’s.
The result: both Americans’ wallets and Americans’ guts suffer.

Catch me on tour with Red Team Blues in Hay-on-Wye, Oxford, Manchester, Nottingham, London, and Berlin!

If you’d like an essay-formatted version of this post to read or share, here’s a link to it on pluralistic.net, my surveillance-free, ad-free, tracker-free blog:
https://pluralistic.net/2023/05/29/oh-shit/#rebyota
[Image ID: A poop emoji wearing a top hat and a monocle, posed against a backdrop of e coli bacteria seen through a high-resolution microscope.]
#pluralistic#stool bank#eat shit and live#pharma#fda#regulatory capture#fecal transplants#microbiomes#rebyota#openbiome#c diff#fmt#fecal microbiota transplant
272 notes
·
View notes
Link
Are you searching for the best food safety consulting companies? If so, look no further than GHP Consulting and Testing LLC. We also help with chemical testing, microbiological testing, calibration of equipment, etc. See it here. https://ghpconsultingandtesting.com/

0 notes
Last Seen Blogs




