#us fda 510 k clearance
Text
https://operonstrategist.com/overcome-fda-510k-clearance-barriers/?utm_source=Social+bookmarking&utm_medium=Off+Page+&utm_campaign=Social+bookmarking
How to overcome FDA 510k Clearance barriers?
#fda 510 k clearance#us fda 510 k clearance#510k fda certificate#510k medical device#510k premarket notification#fda 510k#510k clearance#FDA 510k clearance barriers
0 notes
Text
#510(k) Application#Refusal to Accept#Medical Device#510(k) Clearance#US FDA#510(K)#510(k) submissions#RTA#Regulations
0 notes
Link
#rune labs#fda#apple#apple watch#parkinsons#neuroscience#neurodegenerative disorders#neurodegenerative diseases#brain#big tech#medtech#digital health#health tech#smartwatches#smartwatch
3 notes
·
View notes
Text
FDA 510k — An Overview
FDA 510k is a file containing sufficient information about a device to demonstrate that the medical device is at least as safe and effective as legally marketed devices that are not subjected to PMA. Organizations planning to launch Class I, II, and III devices in the United States intended for human use must submit a 510(k) if pre-market approval is not required.
Most class 1 devices are exempt from 510k requirements. During the review of the file, if the FDA finds the device to be substantially equivalent, it will grant the FDA 510k clearance for medical devices with a ‘(k)’ number
We offer technical and scientific assistance in identifying a suitable predicate device, regulation number, and device code, along with file drafting, e-copy conversion, and submission through the US Agent service. Few manufacturers opt for Q submission before final submission.
Different Types of 510k
Traditional 510k is an original submission that normally has to be provided for the medical device, that requires 510k clearance according to 21 CFR 807. It can also be used to submit if there is any change in the previously cleared device. It generally takes 90 days for the 510k submission.
Abbreviated 510k submission is appropriate when FDA guidance exists for specific medical devices for the demonstration of compliance and any voluntary FDA-recognized consensus standards are available. It covers traditional, as well as a brief report on the usage of FDA guidance documents and FDA-approved standards.
Special 510k submission is for an already cleared device with some changes that don’t require the full review but only the summary document. These changes include indications of use, design, and labelling, but they should not affect the safety or performance of the new, modified device, and well-established methods.
1 note
·
View note
Text
The Rise of Tech-Driven Therapies: Reshaping the Global Chronic Smell and Flavor Loss Treatment Market
The global Chronic Smell And Flavour Market in 2023 is US$ 3146 million, with a compound annual growth rate (CAGR) of 6.4%. Healthcare technology advancements are expected to drive the market to US$ 5850.2 million by 2033.
Over the course of the projection period, the market for chronic smell and flavor loss is anticipated to develop due to an increase in cases of post-COVID-19 flavor loss. In the long run, the market for treatments for chronic smell and flavor loss is anticipated to be impacted by increased awareness brought about by the current COVID-19 pandemic and the anticipated success of the only pipeline candidate.
Preview Next-Level Insights Sample :
https://www.futuremarketinsights.com/reports/sample/rep-gb-16370
The creation of treatments tailored to individual diseases for persistent loss of flavor and odor will be beneficial for market expansion. For example, phosphodiesterase (PDE) inhibitors designed for intranasal delivery are included in CYR-064 formulations. All disorders causing loss of flavor and smell share a common main mechanism. It includes blocking the activation of stem cells, which is often brought about by adenylyl cyclase III acting through cAMP and cGMP.
North America dominates the chronic smell and flavour loss treatment market due to the presence of key market players’ presence, along with recent product launches and established healthcare infrastructure, which will accelerate the market’s growth in the region. The significant R&D investments in Europe abode well for market growth.
The manufacturers are involved in the production of chronic smell and flavor loss treatment in a larger capacity. Research and innovation are also conducted to launch innovative products for chronic smell and flavor loss treatment.
Key Takeaways from the Market Study
As of 2023, the chronic smell and flavor loss treatment market was valued at US$ 3146 Million
From 2023 to 2033, the chronic smell and flavor loss treatment industry is poised to grow at an 6.4% CAGR
By 2033, the chronic smell and flavor loss treatment market is slated to reach a valuation of US$ 5850.2 Million
China is poised to yield a CAGR of 6.0% with respect to chronic smell and flavor loss treatment in 2033
“Growing healthcare spending and increase in the prevalence of smell disorders and taste disorders are expected to radically transform the chronic smell and flavor loss treatment market in the coming years,” comments an analyst at FMI.
Competitive Landscape:
Some of the key players in the chronic smell and flavour loss treatment market like Cyrano Therapeutics, Johns Hopkins, Boys Town National Research Hospitals, MSD Manual, Alcon, Stanford Medicine, Immunomic Therapeutics, Siemens Healthcare Private Limited, Omega Diagnostics Group PLC, HYCOR Biomedical, Inc. and others.
The manufacturers are involved in the production of chronic smell and flavour loss treatment in a larger capacity. Research and innovation are also conducted to launch innovative products for chronic smell and flavour loss treatment.
Cyrano Therapeutics has developed a nasal spray to treat and restore the sense of smell and flavor in those who have experienced a chronic loss of those senses.The Company was founded by an experienced healthcare executive after personally experiencing a chronic loss of smell and flavor. The nasal spray has restored his smell and flavor function and he is now leading a mission to bring it to others.
Hycor Biomedical, a leading manufacturer of in vitro diagnostic products for allergy and autoimmune testing, announced today that it has received U.S. Food and Drug Administration (FDA) 510(k) clearance for its new allergy testing system, NOVEOS.
Key Segments Covered in Chronic Smell and Flavor Loss Treatment Industry Survey
Chronic Smell and Flavor Loss Treatment Market by Classification:
Smell Disorder
Taste disorder
Chronic Smell and Flavor Loss Treatment Market by Symptoms:
Anosmia or Hyposmia
Dysgeusia
Ageusia
Chronic Smell and Flavor Loss Treatment Market by Taste Type:
Self-Assessment Test
Psychophysical Assessment Test
Imaging Test
Other Tests
Chronic Smell and Flavor Loss Treatment Market by Region:
North America
Latin America
Western Europe
Eastern Europe
Asia Pacific
Middle East and Africa(MEA)
0 notes
Text
ReVitalMed: Pioneering Single-Use Medical Device Regeneration
Introduction to Single Use Devices
Single-use medical devices (SUDs) are designed and labeled by their manufacturers to be discarded after one use on a single patient. Common types of SUDs include biopsy forceps, cardiac catheters, drug-eluting stents, and surgical instruments. SUDs offer convenience for healthcare facilities by eliminating cleaning, disinfection, sterilization, and repair costs associated with reusable devices. However, SUDs also generate a significant amount of medical waste.
The Process of Reprocessing SUDs
Some healthcare facilities have begun reprocessing select SUDs to reduce costs and waste. The reprocessing of SUDs involves cleaning, disinfection, sterilization, inspection, and repackaging to prepare a used device for another procedure. The main steps in reprocessing SUDs are:
Cleaning
Thorough cleaning is the first and most important step to remove contaminants from the last clinical use. Robotic, ultrasonic, and manual cleaning methods are used with soap solutions to flush out all residues from lumens and crevices of devices.
Disinfection
After cleaning, high-level disinfection using liquid chemicals like peracetic acid or hydrogen peroxide vapor helps eliminate infectious pathogens and microbes. Strict adherence to disinfectant contact times is necessary for effectiveness.
Inspection and Function Testing
Reprocessed devices then undergo 100% visual, optical, and electronic inspection using magnifying tools. Functionality of moving parts, electrical components, and material integrity are evaluated to prevent failures.
Sterilization
Steam sterilization in an autoclave is the standard method to sterilize heat-stable reprocessed SUDs. Other techniques like ethylene oxide gas or plasma sterilization can be used as well if validated.
Repackaging and Labeling
Once sterilized, devices are individually wrapped or placed in dedicated trays with internal sterilization indicators. Clear labeling denotes the device as reprocessed. Quality control records trace each device back to its original manufacturer.
Regulatory Considerations around SUD Reprocessing
The reprocessing of SUDs raises concerns from a regulatory perspective:
Lack of Manufacturer Validation
Original equipment manufacturers did not design or validate SUDs to withstand multiple uses, high-temperature sterilization, or cleaning chemicals. Significant post-market validation is required.
Liability and Safety Issues
If a reprocessed device malfunctions or causes harm, liability is difficult to determine between the reprocessor and original manufacturer. Safety and performance could be compromised without oversight.
Regulatory Approval Requirements
In many countries like the United States, reprocessors must obtain 510(k) marketing clearance from regulators like the FDA before commercially distributing reprocessed SUDs. However, regulations are still evolving.
Cost-Effectiveness Analysis
While initial reprocessing costs can be high, long-term savings from avoiding purchase of new SUDs can offset cleaning and sterilization fees—especially for high-cost devices. Reliability and remaining useful life need evaluation.
Adoption Challenges in Healthcare Systems
Changing standards of care and ingrained clinical preferences favor single-use over reprocessed devices. Reprocessors must demonstrate comparable quality and safety to gain broader acceptance.
The Future of SUD Reprocessing
As healthcare costs continue rising, SUD reprocessing offers a means to improve sustainability and access to care. However, unresolved concerns around validation, quality assurance, and regulator oversight could slow further development. Standardized best practices, comprehensive regulations, and robust post-market surveillance may encourage more healthcare providers and patients to embrace this option in the future. With appropriate safeguards and diligence by all stakeholders, reprocessing select SUDs shows promise as a practical solution to reduce medical waste and contain costs from expensive disposable technologies.
0 notes
Text

US FDA’s 510(k) Submission & Review Process
510(k) submission is one of the essential steps of getting medical devices approved in the United States. A 510(k) is an FDA regulatory pathway for medical device clearance.
It's crucial as it allows manufacturers to demonstrate that their new device is substantially equivalent to an existing one already on the market, expediting approval.
This process promotes innovation, reduces development costs, and ensures patient safety by evaluating similarities and differences between devices.
Before you start your 510(k) filing, you must consider the below-mentioned factors.
In case you wish to get support for 510(k) submission, you must check out our 510(k) submission consulting services.
1 note
·
View note
Text
Remsleep Holdings Inc.'s DeltaWave Mask has received questions from FDA regarding 510k submission (K233415)
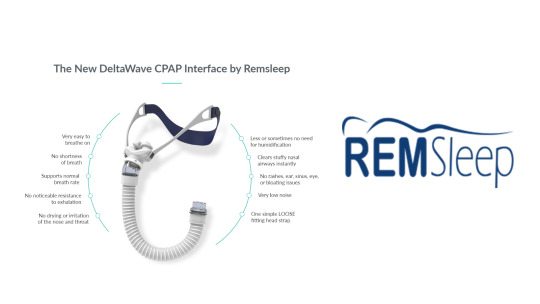
Tampa, FL, January 22, 2024 - RemSleep Holdings Inc. (OTCQB: RMSL), a medical device manufacturer dedicated to forever changing the level of treatment provided to Obstructive Sleep Apnea (OSA) has received a response to the company’s 510(K) submission K233415. The agency has determined that additional information is required. The agency has asked for additional information regarding Administrative Information, Device Description, Substantial Equivalence (SE), Labeling, Biocompatibility, and Performance testing. All these questions can be answered, and we are in the process of responding with the answers.
The agency has also requested additional Shelf-Life testing for DeltaWave packaging. We are in the process of initiating accelerated Shelf-Life testing for the DeltaWave packaging. This request was a surprise to us, but we will promptly comply.
Remsleep CEO, Tom Wood, commented: “Investors want a 510(K) almost as much as Remsleep. We understand the frustration. Investors can be assured we will answer the questions from the agency, perform the additional tests required, and we will make every effort to gain approval for 510(K) submission (K233415) as soon as possible”.
Judy Strzepek comments: “Remsleep submitted our 510(K) on 10/10/2023. Remsleep received feedback from FDA with a request for additional information. This is not unusual. The type of information requested varies based on the type of product. CPAP device and the mask with accessories needed for sleep apnea therapy is a class II device that requires extensive data and information. So, the agency’s request for clarification of any part of the submission is not unexpected. We are working diligently to address any of FDA’s questions. We will do this as quickly as possible, while ensuring our response is thorough and complete.”
For the years 2022 and 2023, FDA 510(K) clearance for products of this category took the following lengths of time from time of submission received by FDA until FDA clearance decision:
Major Competitor 8 Months
Major Competitor 7 Months
Major Competitor 1 year
Major Competitor 6 Months
Major Competitor 9 Months
Major Competitor 10 Months
This information is from an FDA MDUF commitment letter FDA has published on their website. FDA makes these commitments to congress for turnaround of submissions. However, it does not state the fact that each time the FDA asks a question, their review clock stops and does not begin again until the company submits a response. So, while 124 calendar days for 2024 sounds good, we don’t know the actual start date (when company submits 510(K) until FDA gives a decision.
Remsleep has applied for a government grant to perform clinical trials once the 510(K) submission has been approved. The company feels it has a strong chance of being awarded a grant. We will keep investors informed of progress.
Remsleep has been allowed a new US patent for the DeltaWave product(s). The issuance fees have been paid and the new patent is expected to be issued in approximately 7 weeks.
RemSleep will continue to update investors as information becomes available and will be confirmed on the company website: www.remsleep.com; and through the company Twitter feed: @RemsleepInc.
About RemSleep Holdings Inc.
RemSleep Holdings Inc. is a medical device manufacturer dedicated to forever changing the level of treatment provided to obstructive Sleep Apnea patients. Our focus is primarily designing and manufacturing devices and products for the treatment of Sleep Apnea and other respiratory conditions. With over 30 years of collective experience in CPAP therapy, the RemSleep team has extensive knowledge and understanding of CPAP and the challenges of patient compliance. We diligently strive for our products to make the difference and improve the condition of those suffering from Sleep Apnea. www.remsleep.com
https://twitter.com/RemsleepInc
Forward-Looking Statements
Some statements in this release may contain forward-looking information. All statements, other than of historical fact, that address activities, events or developments that the Company believes, expects or anticipates will or may occur in the future (including, without limitation, statements regarding potential acquisitions and financings) are forward-looking statements. Forward-looking statements are generally identifiable by use of the words "may", "will", "should", "continue", "expect", "anticipate", "estimate", "believe", "intend", "plan" or "project" or the negative of these words or other variations on these words or comparable terminology. Forward-looking statements are subject to a number of risks and uncertainties, many of which are beyond the Company's ability to control or predict, that may cause the actual results of the Company to differ materially from those discussed in the forward-looking statements. Factors that could cause actual results or events to differ materially from current expectations include, among other things, without limitation, the inability of the Company to obtain sufficient financing to execute the Company's business plan; competition; regulation and anticipated and unanticipated costs and delays, and other risks disclosed in the Company's public disclosure record on file with the relevant securities regulatory authorities. No information in this press release should be construed as any indication whatsoever of the Company's future revenues, results of operations or stock price. Although the Company has attempted to identify important factors that could cause actual results or events to differ materially from those described in forward-looking statements, there may be other factors that cause results or events not to be as anticipated, estimated or intended. Readers should not place undue reliance on forward-looking statements. The forward-looking statements included in this news release are made as of the date of this news release and the Company does not undertake an obligation to publicly update such forward-looking statements to reflect new information, subsequent events or otherwise unless required by applicable securities legislation.
Investor Relations Contact:
Preya Narain
[email protected]
SOURCE: RemSleep Holdings Inc.
#press release#prism mediawire#stock market#investing#otc markets#otc#prismdigitalmedia#prismmarketview#healthcare#OTCQB#medical devices#Healthcare#RMSL#remsleep
0 notes
Text
Understanding Regulatory Requirements for Sterilization Equipment Market
The regulatory landscape for sterilization equipment market is complex and varies across regions. Regulatory requirements are in place to ensure the safety and efficacy of sterilization processes, particularly in industries such as healthcare and pharmaceuticals.

Buy Full Report for More Insights into the Sterilization Equipment Market Forecast
Download A Free Report Sample
Below is a general overview of the key aspects of regulatory requirements for sterilization equipment:
United States (U.S.):
FDA (Food and Drug Administration):
In the U.S., the FDA regulates sterilization equipment as medical devices. The FDA classifies medical devices into different classes based on risk, and the regulatory pathway varies accordingly.
Sterilization equipment manufacturers must adhere to the Quality System Regulation (QSR) outlined in 21 CFR Part 820, which covers good manufacturing practices.
510(k) Premarket Notification or Premarket Approval (PMA):
Depending on the class of the device, manufacturers may need to submit a 510(k) premarket notification or a PMA application for FDA clearance before marketing their sterilization equipment.
European Union (EU):
CE Marking:
Sterilization equipment must bear the CE marking to indicate compliance with European Union directives. The Medical Devices Regulation (MDR) and In Vitro Diagnostic Devices Regulation (IVDR) outline the regulatory requirements for medical devices, including sterilization equipment.
Conformity Assessment Procedures:
Manufacturers must undergo conformity assessment procedures to demonstrate compliance with essential requirements, including safety and performance. Depending on the device's class, this may involve self-assessment or involvement of a notified body.
International Standards:
ISO Standards:
Compliance with international standards, particularly those developed by the International Organization for Standardization (ISO), is often essential. ISO 13485 specifies requirements for a quality management system for medical devices.
ISO 14971:
Manufacturers are often required to perform risk management according to ISO 14971 to identify and mitigate potential risks associated with their sterilization equipment.
Other Considerations:
Validation and Verification:
Manufacturers must conduct validation and verification activities to ensure the effectiveness and reliability of their sterilization processes. This may include microbial validation studies and performance testing.
Labeling and Instructions for Use:
Accurate and comprehensive labeling, including instructions for use, is crucial for regulatory compliance. Clear instructions help users understand how to operate the sterilization equipment safely and effectively.
Post-Market Surveillance:
Ongoing post-market surveillance is essential for monitoring the safety and performance of sterilization equipment after it has been placed on the market. Manufacturers are typically required to report adverse events.
Good Manufacturing Practices (GMP):
Compliance with GMP principles is a fundamental requirement. Sterilization equipment manufacturers must implement and maintain procedures that ensure the consistent production of safe and effective devices.
Local Requirements:
In addition to global regulations, manufacturers must be aware of and comply with local regulations and requirements in the countries where they intend to market their sterilization equipment.
It's important for manufacturers to stay informed about changes in regulations, standards, and guidance documents relevant to sterilization equipment. Working with regulatory affairs professionals and consultants can help navigate the complex regulatory landscape and ensure compliance with applicable requirements.
0 notes
Text
Ensuring a Smooth Surgical Journey: The Preoperative Assessment Process
PrecisionOS has achieved a significant milestone with the receipt of a 510(k) clearance from the U.S. Food and Drug Administration (FDA) to introduce PrecisionOS Invision™, a cutting-edge patient-specific planning tool utilizing Virtual Reality (VR). Set to be available in early 2022, this patented software empowers surgeons to perform preoperative planning and assessment with the immersive experience of an Oculus Quest 2 device. This revolutionary approach allows surgeons to use the Oculus Quest 2 to interact with, manipulate, and focus on specific anatomical areas before embarking on the actual surgical procedure. PrecisionOS Invision is ushering in a new era of surgical precision, efficiency, and confidence. Get ready to experience surgery in an entirely new dimension.https://www.precisionostech.com/preoperative-planning/
0 notes
Text
What is 510k and its clearance process?
What is 510k and its clearance process?
510k approval is a process to get marketing clearance for a Class II Medical Device. Class II Medical Devices are non-invasive devices usually meant for secondary treatment and aid. 510k is also referred to as Pre-Market Notification.
The process of getting a 510k clearance requires finding an equivalent product (predicate device) to a new product to prove its substantial equivalence. Once the device is proven equivalent to an already approved equivalent it can be legally marketed as per US FDA guidelines.
510k submission checklist
The entire process of the 510k is divided into two phases:
(I) Acceptance Review
Acceptance review takes up to 15 days. In case, the 510k is not accepted at the primary level, RTA is issued. RTA refers to Refuse to Accept. In such a case some more documentation is needed to validate the product.
(III)Substantive Review
The substantive review takes up to 60 days. At this stage, the approval body may ask for additional information to justify the indicated use of the product.
What does FDA require as additional information?
The additional information, in general, has those facts that completely justify the intended use or impact of the product.
Additional information requests include:
a. Improper device description
b. Discrepancies in the indications for use or device description
c. Data related to indicated use
d. Inability to comply with set standards or regulations
e. Non-satisfactory clinical data
Operon Strategist is medical device consultancy. which provide the consultation on FDA 510k clearance process
#fda 510 k clearance#usfda 510k#510k fda certificate#510k medical device#510k database#us fda 510 k clearance#fda registered 510 k medical device#510k premarket notification#fda 510k#510k consultants
0 notes
Text
#Medical Devices#Post-approval Changes#510(k) Clearance#US FDA#Regulations#510(K)#510(k) submissions#post-approval change management#medical device products
0 notes
Text
US: Amazon's virtual health clinic is now equipped to provide treatment for symptoms related to a cough, cold, or flu

- By InnoNurse Staff -
Amazon recently announced that its virtual health care marketplace, Amazon Clinic, can now treat patients for a cough, cold or flu.
Read more at TechCrunch
///
Other recent news and insights
Denti.AI has received FDA 510(k) clearance for Denti.AI Detect, an AI-powered imaging solution, which enhances disease detection in both intra- and extraoral dental radiography and streamlines charting automation (Denti.AI/PRNewswire)
Bangladesh: Digital health startup Arogga closes $5.5M round (Tech Funding News)
#amazon#big tech#digital health#telehealth#health tech#primary care#pharmacy#dentiai#arogga#usa#bangladesh#dentistry#imaging#ai
0 notes
Text
PCR/RT Market Growth Opportunities & Limitations
PCR/RT Market, Technology (Conventional, qPCR, dPCR), Product (Instrument, Reagents, Software), Application (Genotyping, Sequencing, Gene expression, diagnostics), Enduser (Academia, pharma-biotech, applied) and Geography (North America, Europe, Asia-Pacific, Middle East and Africa and South America)
Market Overview
The global PCR/RT market size is projected to reach USD 11.5 billion by 2026 at a CAGR of 6.7% from USD 7.5 billion in 2021 during the forecast period 2021-2028.
PCR or polymerase chain reaction is a widely used method for making multiple copies of a specific DNA sample. This is used in order to amplify the content of the desired segment of a DNA in a cell free medium. It was first invented in 1983 by Kary Mullis. The DNA polymerase enzyme used in the process is Taq polymerase as it is a thermostable polymerase.

With the surge in case of infectious disease and genetic disorders along with the recent development of PCR technologies and investments in the sector are some of the factors that have supported long-term expansion for PCR/RT Market.
The COVID-19 pandemic is causing widespread concern and economic hardship for consumers, businesses, and communities across the globe. But the market of PCR/RT technology witnessed a boom during the pandemic as PCR is a standers test for gene sequencing and became an ideal tool for the lab testing for COVID-19
To Understand Business Strategies, Request for a Sample Report at: https://www.delvens.com/get-free-sample/pcr-market-trends-forecast-till-2028
Regional Analysis
Asia Pacific is expected to be the largest market during the forecast period. The advancement in the healthcare system along with the increased research and development in the sector have led to growth in the PCR technology in the region.
Competitive Landscape
Key Players
Thermo Fisher Scientific, Inc
F. Hoffman-La Roche Ltd
Bio-Rad Laboratories, Inc
QIAGEN N.V.
Takara Bio, Inc.
Agilent Technologies, Inc.
bioMérieux S.A.
Fluidigm Corporation
Danaher Corporation
Abbott Laboratories
Merck KGaA
Becton Dickinson and Company
Promega Corporation
Eppendorf AG
Analytik Jena AG
To Grow Your Business Revenue, Make an Inquiry Before Buying at: https://www.delvens.com/Inquire-before-buying/pcr-market-trends-forecast-till-2028
Recent Developments
In 2020, FDA 510(k) clearance was given to Roche for the cobas BKV Test on the cobas 6800 and 8800 Systems
In 2020, Roche’s cobas SARS-CoV-2 & Influenza A/B Test received emergency use authorization for use on the cobas 6800/8800 Systems from the FDA
Reasons to Acquire
Increase your understanding of the market for identifying the best and suitable strategies and decisions on the basis of sales or revenue fluctuations in terms of volume and value, distribution chain analysis, market trends and factors
Gain authentic and granular data access for PCR/RT market so as to understand the trends and the factors involved behind changing market situations
Qualitative and quantitative data utilization to discover arrays of future growth from the market trends of leaders to market visionaries and then recognize the significant areas to compete in the future
In-depth analysis of the changing trends of the market by visualizing the historic and forecast year growth patterns
Get Direct Order of this Report: https://www.delvens.com/checkout/pcr-market-trends-forecast-till-2028
Report Scope
PCR/RT Market is segmented into Technique, Product, Application, End User and geography.
On the basis of Technique
Conventional PCR
Real-time PCR (qPCR)
Digital PCR (dPCR)
Reverse Transcription PCR (RT-PCR)
Hot-start PCR
Multiplex PCR
Other PCR
On the basis of End-User
Hospitals and diagnostic labs
Healthcare industry
Academia and government organizations
Pharma-biotech companies
Applied industries
Other end users
On the basis of Application
Gene expression analysis
Genetic sequencing
Genotyping
Nucleic acid detection
Nucleic acid synthesis
Standard validation/verification
Diagnostic Applications
Environmental Applications
Other Applications
On the basis of Product
Instruments
Reagents and consumables
Software and services
On the basis of Region
Asia Pacific
North America
Europe
South America
Middle East & Africa
About Us:
Delvens is a strategic advisory and consulting company headquartered in New Delhi, India. The company holds expertise in providing syndicated research reports, customized research reports and consulting services. Delvens qualitative and quantitative data is highly utilized by each level from niche to major markets, serving more than 1K prominent companies by assuring to provide the information on country, regional and global business environment. We have a database for more than 45 industries in more than 115+ major countries globally.
Delvens database assists the clients by providing in-depth information in crucial business decisions. Delvens offers significant facts and figures across various industries namely Healthcare, IT & Telecom, Chemicals & Materials, Semiconductor & Electronics, Energy, Pharmaceutical, Consumer Goods & Services, Food & Beverages. Our company provides an exhaustive and comprehensive understanding of the business environment.
Contact Us:
UNIT NO. 2126, TOWER B,
21ST FLOOR ALPHATHUM
SECTOR 90 NOIDA 201305, IN
+44-20-8638-5055
The PCR/RT Market report answers a number of crucial questions, including:
Which companies dominate the PCR/RT Market?
What current trends will influence the market over the next few years?
What are the market's opportunities, obstacles, and driving forces?
What predictions for the future can help with strategic decision-making?
What advantages does market research offer businesses?
Which particular market segments should industry players focus on in order to take advantage of the most recent technical advancements?
What is the anticipated growth rate for the market economy globally?
0 notes
Text
The Rise of Tech-Driven Therapies: Reshaping the Global disposable endoscope market
The disposable endoscope market is expected to have grown to a valuation of US$ 1.3 billion, with a compound annual growth rate (CAGR) of 17.7% from 2023 to 2033. According to projections, the global market is expected to be worth US$ 6.8 billion by 2033.
Disposable endoscopes are beginning to provide a simple solution to the global problem of surgical site infections. The availability of various medical instruments, such as laryngoscopes, proctoscopes, hysteroscopes, esophagogoscopes, bronchoscopes, and cystoscopes, may propel the global market.
Discover Industry Secrets Through Your Sample Report:
https://www.futuremarketinsights.com/reports/sample/rep-gb-15535
Combating Hospital Infections: Disposable Endoscopes Offer a Safe and Effective Solution
The global disposable endoscopes market is experiencing a surge in growth, driven by a critical need for safer surgical practices. Disposable endoscopes are revolutionizing endoscopy by reducing the risk of Hospital Acquired Infections (HAIs) associated with reusable endoscopes.
Multiple Factors Propelling Market Growth:
Several factors are contributing to the significant rise of the disposable endoscopes market:
Enhanced Patient Safety: Disposable endoscopes eliminate the risk of cross-contamination often associated with reprocessed reusable endoscopes, leading to improved patient safety outcomes.
Minimally Invasive Advantage: The growing preference for minimally invasive surgeries, which offer faster recovery times and reduced patient discomfort, is driving the demand for disposable endoscopes specifically designed for these procedures.
Chronic Disease Diagnosis: The rising prevalence of chronic diseases like cancer, inflammatory bowel disease, and respiratory illnesses is fueling the use of endoscopes for diagnosis and treatment, creating a larger market for disposable options.
Key Takeaways:
The global disposable endoscopes market is estimated to reach US$6.8 billion by 2033, reflecting a significant rise from US$1.3 billion in 2023.
This growth is projected at a remarkable compound annual growth rate (CAGR) of 17.7% throughout the forecast period.
The increasing adoption of minimally invasive procedures and the rising prevalence of chronic diseases are key drivers for market expansion.
Competitive Landscape:
The market is highly fragmented, with several competitors in the disposable endoscope production sphere. To meet the rising consumer demand and expand their customer base, these companies are implementing various strategies such as mergers & acquisitions, partnerships, collaborations, and new product launches.
Recent Industry Developments
In July 2022, Ambu Inc. received 510(k) regulatory clearance from the USA Food and Drug Administration (FDA) for its Ambu aScope 5 Broncho fifth-generation sterile bronchoscopes.
In May 2021, the PENTAX Medical ONE Pulmo, a brand-new single-use bronchoscope from PENTAX Medical Europe, bagged the CE mark. This ground-breaking product offers pulmonary treatment of the utmost quality. It is a disposable bronchoscope with enhanced suction power and high-resolution images.
Key Companies Profiled:
Ambu A/S
EndoLook
Medtronic Plc
HOYA Corporation
Flexicare Medical Limited
Disposable Endoscopes Market Segmentation:
By Product:
Bronchoscopes
Cystoscopes
Duodenoscopes
Esophagoscopes
Hysteroscopes
Laryngoscopes
Proctoscopes
Ureteroscopes
By End User:
Hospitals
Ambulatory Surgical Center
Diagnostic Center
By Region:
North America
Latin America
Europe
East Asia
South Asia
Oceania
The Middle East & Africa (MEA)
0 notes
Text
0 notes
Last Seen Blogs

sugar-bi-scuit
sugar-bi-scuit

lovehunter666
Holla

kpthomas
hold me wrap me unfold me

bu0nanotte
bitches want paté? then paté they shall have.

10-salem-19
FootBaller